
No olvides visitar la barra lateral, y estudia los Diplomados Online que RELAPED tiene para ti.


Blog oficial de RELAPED para la publicación de artículos clínicos, casos, revisiones de investigación y acceso a revistas y libros virtuales. Diseñado por médicos y enfermeros y dirigido exclusivamente para profesionales de la salud.
No olvides visitar la barra lateral, y estudia los Diplomados Online que RELAPED tiene para ti.
, , , et al. Immune profile of adipose tissue from youth with obesity and asthma. Pediatric Obesity. 2023;e13078. doi:10.1111/ijpo.13078
INTRODUCCIÓN
La otitis media aguda (OMA) es un problema común en niños y representa una gran proporción de las prescripciones de antibióticos pediátricos.
TERMINOLOGÍA
- Efusión del oído medio (EOM)*: Líquido en la cavidad del oído medio; la EOM ocurre tanto en OMA como en otitis media con efusión (OME).
- Otitis media aguda*: Infección bacteriana aguda del líquido del oído medio; también llamada otitis media supurativa.
- Otitis media con efusión (OME)*: Líquido en el oído medio que no está infectado; también llamado otitis media serosa, secretoria o no supurativa.
La OMA y la OME son parte de un espectro continuo; la OME frecuentemente precede al desarrollo de OMA o sigue a su resolución.
ANATOMÍA DEL OÍDO MEDIO
El oído medio es una cavidad entre el canal auditivo externo y el oído interno que contiene la cadena osicular (figura 1). El nervio facial atraviesa la pared medial del oído medio. El oído medio es contiguo con la trompa de Eustaquio, ático (epitímpano) y células aéreas mastoideas.

La membrana timpánica está dividida en cuatro cuadrantes: una línea trazada a lo largo del manubrio del martillo divide los cuadrantes anterior y posterior, y una línea a través del umbo divide los cuadrantes superior e inferior (figura 2). La pars flaccida es la pequeña porción de la membrana timpánica por encima del proceso lateral del martillo; es más gruesa y menos tensa que el resto de la membrana timpánica. El resto de la membrana timpánica (la pars tensa) es más delgada que la pars flaccida y está suspendida del anillo timpánico fibroso.

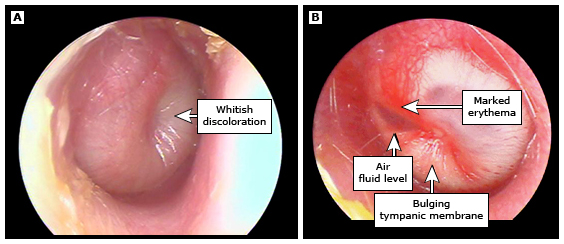
El oído medio normal está aireado y la membrana timpánica normal está intacta, ligeramente convexa, translúcida y móvil (imagen 1).

PRESENTACIÓN CLÍNICA
Otitis media aguda:
Los síntomas de OMA en niños incluyen dolor de oído, frotamiento del oído, pérdida de audición y drenaje del oído. La fiebre ocurre en un tercio a dos tercios de los niños con OMA, aunque una temperatura >40°C (104°F) es inusual sin bacteriemia u otro foco de infección.
El dolor de oído es la queja más común y el mejor predictor de OMA. Sin embargo, el dolor de oído y otros síntomas relacionados con el oído no siempre están presentes. En un estudio prospectivo de 335 episodios consecutivos de OMA, el dolor de oído estuvo ausente en el 17 por ciento. La OMA sin quejas de dolor de oído ocurrió con más frecuencia en niños menores de dos años (que pueden no ser capaces de quejarse de dolor de oído) que en niños mayores (25 versus 7 por ciento). Otras causas de dolor de oído, pérdida de audición y otorrea en niños se discuten por separado.
Los niños pequeños con OMA, especialmente los lactantes, pueden presentar síntomas y signos inespecíficos (por ejemplo, fiebre, irritabilidad, sueño alterado o inquieto, mala alimentación/anorexia, vómitos, diarrea). Los síntomas de OMA pueden solaparse con los de la infección del tracto respiratorio superior sin OMA o pueden ser sutiles o ausentes. La falta de especificidad de los síntomas en niños pequeños hace que el diagnóstico sea desafiante y subraya la importancia de la otoscopia, que también es desafiante en niños pequeños.
Complicaciones de la OMA:
Los niños con complicaciones de OMA pueden presentar hallazgos relacionados con la complicación, incluyendo:
- Hinchazón postauricular y protrusión del pabellón auditivo (imagen 2A-B) son característicos de mastoiditis.


- Parálisis de nervios craneales (por ejemplo, nervio facial, nervio abducens) pueden estar relacionados con mastoiditis aguda, petrositis, colesteatoma o complicaciones intracraneales.
- Signos meníngeos, déficits de nervios craneales y/o hallazgos neurológicos focales pueden estar relacionados con complicaciones intracraneales (por ejemplo, meningitis, absceso cerebral, absceso epidural o subdural, trombosis del seno lateral o cavernoso). Estas complicaciones se discuten en revisiones temáticas individuales.
EVALUACIÓN OTOSCÓPICA
La evaluación otoscópica es necesaria para el diagnóstico de OMA.
Procedimiento:
- Eliminación de cerumen: El cerumen obstructivo debe ser eliminado del canal externo para asegurar una visión clara de la membrana timpánica. El cerumen se elimina de manera más práctica y conveniente bajo visión directa.
- Equipo:
- Un otoscopio con una fuente de luz adecuada (por ejemplo, una bombilla halógena con un brillo ≥100 candelas/pie [1000 lux]) y una batería completamente cargada; se prefiere un otoscopio neumático con una cabeza redonda porque proporciona el mejor sello para la otoscopia neumática (imagen 3).

- Espéculos auriculares de varios tamaños; se debe usar el tamaño más grande que se ajuste cómodamente a la parte cartilaginosa del canal auditivo externo (un espéculo de 4 mm funcionará para la mayoría de los niños); el diámetro exterior del espéculo puede aumentarse colocando un pequeño pedazo de tubo de goma alrededor de la punta.
- Bulbo y tubo insuflador; el sistema neumático debe ser revisado periódicamente para detectar fugas ocluyendo la punta del espéculo con un dedo y apretando el bulbo de goma para ver si se siente resistencia.
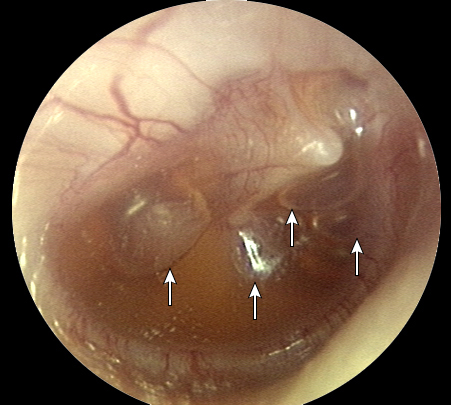
- Evaluación de la membrana timpánica: Cada cuadrante de la membrana timpánica (figura 2) debe ser evaluado sistemáticamente para evaluar posición, movilidad, translucidez, color y otros hallazgos (por ejemplo, niveles aire-líquido, perforación, bolsas de retracción, colesteatoma). La movilidad de la membrana timpánica puede ser evaluada con otoscopia neumática. La evaluación sistemática de la membrana timpánica y la otoscopia neumática son desafiantes, especialmente en niños pequeños que pueden no cooperar con el examen.
- Otoscopia neumática: La otoscopia neumática puede ser dolorosa en niños con OMA. No es necesaria en niños con membranas timpánicas abultadas porque todas las membranas timpánicas abultadas tienen movilidad disminuida o ausente. La otoscopia neumática crea presiones positivas y negativas dentro del canal auditivo externo usando el bulbo insuflador del otoscopio. Se crea una presión positiva cuando se comprime el bulbo insuflador y una presión negativa cuando se libera. Es esencial un sello hermético entre el espéculo y el canal auditivo externo. La movilidad de la membrana timpánica se visualiza mejor en el cuadrante posterosuperior o en la pars flaccida, donde la membrana timpánica es más complaciente (figura 2). La membrana timpánica se aleja del observador con presión positiva y se acerca al observador con presión negativa. La movilidad se describe como normal, ausente, disminuida o aumentada. Cuando hay alta presión negativa en la cavidad del oído medio, la membrana timpánica puede estar máximamente retraída e incapaz de alejarse del observador con presión positiva. La movilidad de una membrana timpánica retraída puede evaluarse creando presión negativa en el canal auditivo externo. El otoscopio debe ser retirado del canal externo y comprimir el bulbo. Después de comprimir el bulbo, se vuelve a insertar el otoscopio en el canal externo. Cuando se asegura el sello, se libera el bulbo, creando una presión negativa, lo que permite que la membrana timpánica retraída se mueva hacia el observador en una posición neutral.
- Otoscopia digital: La otoscopia digital, que está en uso más frecuente desde 2015, incorpora una cámara de alta resolución en el otoscopio o endoscopio para permitir la fotografía o videografía. Hay disponibles varias marcas de otoscopios digitales. Ninguno de los dispositivos permite la otoscopia neumática. Un tipo de dispositivo se conecta a un smartphone para crear un otoscopio digital que proporciona imágenes similares a las que se ven a través de un otoscopio convencional; estos dispositivos típicamente usan espéculos grandes, requieren eliminación de cerumen y pueden ser incómodos de usar ya que el smartphone funciona como el mango del otoscopio. Otro otoscopio digital utiliza un espéculo delgado para eludir o atravesar el cerumen para proporcionar una vista de la membrana timpánica. Las imágenes grabadas pueden ser vistas en la pantalla táctil ampliada del otoscopio o cargadas a una computadora para su ampliación y revisadas por uno o varios observadores (incluidos los cuidadores). Esta es una de las principales ventajas de estos dispositivos que permite la observación concurrente con fines de instrucción y diagnóstico de una imagen "fija" para "estudiar". Los dispositivos otoscópicos digitales abordan uno de los principales desafíos del examen del oído en niños pequeños, que es la documentación. Un estudio ha demostrado una mayor concordancia entre los aprendices y los supervisores cuando se utiliza el otoscopio digital en comparación con el otoscopio tradicional. Si el uso de la otoscopia digital mejorará el diagnóstico de la OMA y reducirá el uso innecesario de antibióticos requiere un estudio adicional. Las habilidades otoscópicas, incluyendo la interpretación precisa de los hallazgos, pueden mejorarse a través del entrenamiento.
Hallazgos de la membrana timpánica:
En niños con OMA, los hallazgos clásicos del examen incluyen un oído medio lleno de líquido y una membrana timpánica que está abultada, opaca, amarilla o blanca y tiene movilidad disminuida o ausente con otoscopia neumática, si se realiza otoscopia neumática. Sin embargo, esta constelación de hallazgos no siempre está presente.


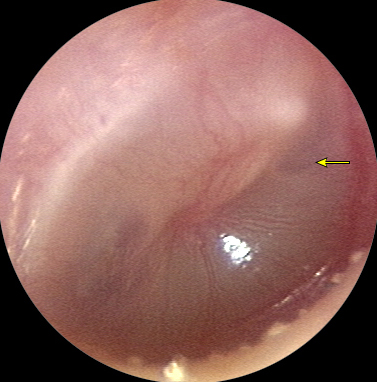
- Membrana timpánica abultada: Una membrana timpánica abultada es la característica distintiva de la OMA y diferencia la OMA de la otitis media con efusión (OME). Una membrana timpánica abultada indica tanto inflamación aguda como efusión del oído medio (EOM, y por lo tanto movilidad disminuida o ausente). El abultamiento es primero aparente en el área posterosuperior, donde la membrana timpánica es más complaciente (figura 2). Cuando la membrana timpánica está abultada, el mango del martillo está oscurecido. La membrana timpánica puede aparecer llena en lugar de abultada cuando hay menores cantidades de líquido infectado en el oído medio. En un gran estudio que correlaciona los hallazgos del examen de otoscopia con los resultados de la miringotomía (como herramienta diagnóstica para la OMA), el valor predictivo de una membrana timpánica abultada osciló entre 83 y 99 por ciento. En otro estudio que correlaciona los hallazgos del examen con el diagnóstico de OMA por otoscopistas experimentados, el 92 por ciento de los niños con OMA tenía una membrana timpánica abultada en comparación con ninguno de los niños con OME o sin efusión. En estos estudios, la OMA era improbable cuando la membrana timpánica estaba en una posición neutral o retraída. Cuando la membrana timpánica está retraída, el mango del martillo parece acortarse y el proceso lateral se vuelve más prominente y parece estar más cerca del otoscopio. La retracción de la membrana timpánica es una característica de la OME.
- Perforación aguda con otorrea purulenta: Una perforación aguda con otorrea purulenta establece el diagnóstico de OMA siempre que se excluya la otitis externa.
- Movilidad disminuida o ausente: La movilidad disminuida o ausente de la membrana timpánica es un signo de MEE, siempre que el sello sea hermético durante la otoscopia neumática. La movilidad disminuida también puede ocurrir con la miringoesclerosis de la membrana timpánica.

- Membrana timpánica nublada u opaca: La membrana timpánica, o una parte de ella, puede aparecer nublada u opaca cuando hay líquido en el oído medio. Este hallazgo no ayuda a diferenciar la OMA de la OME.

- Color de la membrana timpánica: Una membrana timpánica blanca o amarillo pálido indica pus en la cavidad del oído medio, un signo de OMA. El líquido del oído medio que no está infectado, generalmente aparece de color ámbar, gris o azul.
TIMPANOMETRÍA Y REFLECTOMETRÍA ACÚSTICA:
La timpanometría y la reflectometría acústica pueden utilizarse para confirmar la presencia de líquido en el oído medio.
DIAGNÓSTICO:
Se requieren criterios diagnósticos estrictos para distinguir la OMA de la otitis media con efusión (OME). No se puede subestimar la importancia de un diagnóstico preciso de la OMA. Un diagnóstico preciso previene el uso excesivo de antibióticos, lo que lleva al desarrollo de organismos resistentes.
DIAGNÓSTICO DIFERENCIAL:
La principal consideración en el diagnóstico diferencial de la OMA es la otitis media con efusión (OME).
Otitis media con efusión (OME): La efusión del oído medio con movilidad disminuida y opacificación o nubosidad de la membrana timpánica ocurre tanto en la OMA como en la OME. Aunque la distinción entre OMA y OME puede ser difícil porque son parte de un espectro continuo, otros hallazgos otoscópicos pueden ser útiles.

Otras condiciones: Otras condiciones comparten algunas de las características otoscópicas y no otoscópicas de la OMA, pero la historia clínica y el examen físico deberían distinguir fácilmente estas condiciones de la OMA.
- Enrojecimiento de la membrana timpánica: El enrojecimiento de la membrana timpánica puede ser causado por congestión vascular debido al llanto, fiebre alta, infección del tracto respiratorio superior con congestión e inflamación de la mucosa que recubre todo el tracto respiratorio, trauma y/o eliminación del cerumen.
- Movilidad disminuida o ausente de la membrana timpánica: Además de la OMA y la OME, la movilidad disminuida o ausente de la membrana timpánica puede ser causada por la miringoesclerosis y la alta presión negativa en la cavidad del oído medio.
- Dolor de oído: El dolor de oído puede ser causado por otitis externa, trauma en el oído, infecciones de garganta, cuerpos extraños o síndrome de articulación temporomandibular.
**Conclusión**:
El diagnóstico preciso y oportuno de la otitis media aguda es esencial para proporcionar el tratamiento adecuado y evitar complicaciones. La otoscopia sigue siendo la herramienta principal para el diagnóstico, y se deben interpretar correctamente los hallazgos otoscópicos en el contexto de los síntomas clínicos. El uso indebido de antibióticos no solo es ineficaz, sino que también conduce al desarrollo de resistencias bacterianas. Por lo tanto, es crucial diferenciar entre la OMA y otras afecciones del oído medio, como la OME.
REFERENCES
INTRODUCCIÓN
Casi todos los recién nacidos desarrollan niveles elevados de bilirrubina (es decir, bilirrubina sérica o plasmática total [TSB] >1 mg/dL [17 micromol/L], que es el límite superior de la normalidad para los adultos). A medida que aumentan los niveles de bilirrubina, el recién nacido puede desarrollar ictericia visible.
Los recién nacidos con hiperbilirrubinemia grave (definida como TSB >25 mg/dL [428 micromol/L] en recién nacidos a término y prematuros tardíos [edad gestacional ≥35 semanas]) corren el riesgo de desarrollar neurotoxicidad inducida por bilirrubina:
DEFINICIONES
METABOLISMO DE LA BILIRRUBINA
El conocimiento de los pasos básicos del metabolismo de la bilirrubina es esencial para comprender la patogenia de la hiperbilirrubinemia neonatal. El metabolismo de la bilirrubina se revisa brevemente aquí y se discute en detalle por separado (figura 1).

Producción de bilirrubina - La bilirrubina es un producto del catabolismo del hemo. En los recién nacidos, aproximadamente del 80 al 90 por ciento de la bilirrubina se produce durante la descomposición de la hemoglobina de los glóbulos rojos o por eritropoyesis ineficaz. El 10-20% restante procede de la descomposición de otras proteínas que contienen hemo, como los citocromos y la catalasa. Las mediciones de la producción de monóxido de carbono (CO), como los niveles de CO al final de la marea (ETCO) o de carboxihemoglobina (COHb), ambos corregidos para el CO ambiental (ETCOc y COHbc, respectivamente), pueden utilizarse como índices de la producción de bilirrubina in vivo.
La bilirrubina se produce en dos pasos (figura 1).
Aclaramiento y excreción de bilirrubina - El aclaramiento y la excreción de bilirrubina se producen en los siguientes pasos posteriores (figura 1):

Circulación enterohepática - La bilirrubina conjugada secretada no puede ser reabsorbida por las células epiteliales intestinales. En el adulto, las enzimas bacterianas intestinales la reducen a urobilina. Sin embargo, al nacer, el intestino del lactante es estéril y, por consiguiente, los lactantes tienen muchas menos bacterias en el intestino, por lo que muy poca bilirrubina conjugada, si es que hay alguna, se reduce a urobilina. En el lactante, la betaglucuronidasa de la mucosa intestinal desconjuga la bilirrubina conjugada. La bilirrubina no conjugada puede entonces reabsorberse a través de la pared intestinal y reciclarse en la circulación, un proceso conocido como circulación enterohepática de la bilirrubina.
HIPERBILIRRUBINEMIA NEONATAL BENIGNA
La hiperbilirrubinemia neonatal benigna (también denominada anteriormente "ictericia fisiológica") es una bilirrubinemia no conjugada (de reacción indirecta) que se produce en casi todos los recién nacidos [1]. Se trata de un fenómeno transitorio normal causado por el recambio de glóbulos rojos fetales, la inmadurez del hígado del recién nacido para metabolizar eficazmente (conjugar) la bilirrubina y el aumento de la circulación enterohepática.
Los niveles normales de bilirrubina total en suero o plasma (TSB) en adultos son <1 mg/dL, mientras que los recién nacidos a término suelen tener niveles de TSB que alcanzan un pico medio de aproximadamente 8 a 9 mg/dL porque:

Descarga el libro en: DESCARGAR No olvides visitar la barra lateral, y estudia los Diplomados Online que RELAPED tiene para ti.


