
Artículo original de:
Barboza J., et al (2015). Pediatría y Neonatología Clínica. Educación Médica Contínua. Ed. Grafficort. Perú
Cardiología Pediátrica
Joshuan Barboza Meca. MCE
Introducción
Desarrollo de la sangre y del aparato vascular
El desarrollo del aparato vascular empieza en la pared del saco vitelino durante la tercera semana de la gestación (a los 18 días) con la formación de islotes sanguíneos. En este momento el embrión ha alcanzado un tamaño demasiado grande para que el oxígeno llegue a todos los tejidos mediante difusión. Por eso se tienen que desarrollar muy pronto el corazón y el sistema vascular. Como los tejidos que suelen producir células sanguíneas en el adulto no se han empezado a formar, la hematopoyesis en el saco vitelino sirve como adaptación temporal para cubrir las necesidades inmediatas del embrión.
De acuerdo con la hipótesis más extendida, las células fundadoras de los islotes sanguíneos, que se denominan hemangioblastos, tienen una capacidad de desarrollo bipotencial y pueden dar origen tanto a células endoteliales como a células madre hematopoyéticas. Una vez están comprometidas en una de estas dos líneas, las células hijas pierden la capacidad para formar el otro tipo celular. Una hipótesis más reciente postula que cuando los hemangioblastos colonizan el saco vitelino, estas células ya han sido segregadas hacia los linajes hematopoyético y endotelial. Las investigaciones en embriones de ratón sugieren que en vez de formar islotes sanguíneos, las células hematopoyéticas se agregan en una banda sanguínea que rodea el saco vitelino (Figura 1).

Hematopoyesis embrionaria
Las células precursoras hemangiogénicas aparecen por primera vez en el mesodermo posterolateral durante la gastrulación y desde aquí migran a los órganos formadores de sangre iniciales. Bajo la influencia de Runx-1 algunos descendientes siguen el linaje hematopoyético, mientras que otros responden a Hoxa3 y siguen el linaje endotelial. Un tercer grupo sigue otro linaje y terminan formando células de músculo liso vascular. Aunque la formación de la sangre (hematopoyesis) empieza en el saco vitelino, las células derivadas del mismo pronto son sustituidas por otras células sanguíneas, que se originan de forma independiente en otros focos de hematopoyesis.
Los islotes sanguíneos contienen células madre hematopoyéticas pluripotenciales, que pueden originar la mayor parte de los tipos celulares presentes en la sangre embrionaria. Los eritrocitos producidos en el saco vitelino son células nucleadas grandes que penetran en la corriente sanguínea justo antes de que el tubo cardíaco empiece a latir, en torno al día 22 de la gestación. Durante las primeras 6 semanas los eritrocitos circulantes derivan mayoritariamente del saco vitelino, pero en ese momento se están preparando las fases siguientes de la hematopoyesis.
El análisis de embriones humanos ha demostrado que, comenzando en el día 28, la hematopoyesis intraembrionaria definitiva empieza en pequeños agregados de células (acúmulos paraaórticos) en el mesodermo esplacnopleural asociado a la pared ventral de la aorta dorsal, y poco después en la región aorta /cresta genital/ mesonefros (AGM). Los precursores celulares de la región AGM alcanzan por vía sanguínea los lugares formadores de sangre del hígado, del saco vitelino y de la placenta. Las células madres hematopoyéticas formadas en la región AGM, el saco vitelino y en la placenta son transportadas hasta el hígado a través de la circulación de este órgano. Hacia las 5 o 6 semanas de gestación, los focos de hematopoyesis van siendo cada vez más destacados en el hígado.
Tanto en el saco vitelino como en los primeros focos de hematopoyesis embrionaria, las células endoteliales conservan durante un breve período la capacidad de dar origen a células productoras de sangre. En la actualidad, existen evidencias de que en la región AGM la señalización del óxido nítrico, resultante del intenso esfuerzo causado por el flujo sanguíneo en las células endoteliales, puede inducir la transformación en células madre hematopoyéticas.
Los eritrocitos que se producen en el hígado son bastante distintos a los derivados del saco vitelino. Aunque siguen siendo mucho más grandes que las células sanguíneas rojas normales del adulto, los eritrocitos de origen hepático no tienen núcleo y contienen distintos tipos de hemoglobina. Hacia las 6-8 semanas de gestación en el humano, el hígado sustituye al saco vitelino como principal fuente de células sanguíneas. Aunque el hígado sigue produciendo hematíes hasta el período neonatal temprano, su contribución empieza a decaer en el sexto mes de gestación. En este momento, la formación de sangre se desplaza hacia la médula ósea, el lugar definitivo de la hematopoyesis en el adulto. Este desplazamiento está controlado por el cortisol secretado en la corteza suprarrenal fetal. En ausencia de cortisol, la hematopoyesis permanece confinada en el hígado. Antes de que dicha hematopoyesis se establezca por completo en la médula ósea se pueden formar pequeñas cantidades de sangre en el epiplón y posiblemente en el bazo.
Aspectos celulares de la hematopoyesis
Las primeras células madre hematopoyéticas que se originan en el embrión son verdaderamente pluripotenciales, porque pueden dar origen a todos los tipos celulares presentes en la sangre. Estas células madre pluripotenciales, que a veces se denominan hemocitoblastos, tienen gran capacidad proliferativa. Producen gran número de progenitores, la mayor parte de los cuales son células en el siguiente estadio de diferenciación, aunque también producen un pequeño número de células de su tipo pluripotencial original, que actúan como una reserva para reponer distintas líneas individuales de células en caso de necesidad. En fases muy precoces del desarrollo, la línea de células que forman sangre de manera activa se subdivide en dos linajes distintos. Las células madre linfoides producen en última instancia las dos líneas de linfocitos: los linfocitos B (responsables de la producción de anticuerpos) y los linfocitos T (que se encargan de las reacciones inmunes celulares). Las células madre mieloides son precursoras de las otras líneas de células sanguíneas: los eritrocitos, los granulocitos (neutrófilos, eosinófilos y basófilos), los monocitos y las plaquetas. Las células madre de la segunda generación (linfoides y mieloides) son todavía pluripotenciales, aunque su capacidad de desarrollo está restringida, ya que ninguna de ellas puede dar lugar a descendencia del otro tipo (Figura 2).
Según su comportamiento en determinadas situaciones experimentales, las células madre hematopoyéticas se denominan a menudo unidades formadoras de colonias (CFU). Las células madre de primera generación se llaman CFU-ML, porque pueden originar líneas celulares linfoides y mieloides. Las células madre de segunda generación reciben el nombre de CFU-L (linfocitos) y de CFU-S (bazo) (determinadas a partir de experimentos en los que la diferenciación de células madre se estudió en bazos irradiados). En todos los casos la progenie de las CFU-ML y las CFU-S son células madre comprometidas, que sólo pueden dar origen a un tipo de célula sanguínea madura. Los tipos celulares que forman cada linaje deben atravesar varios estadios de diferenciación antes de llegar a su fenotipo maduro.
Existen factores estimuladores de colonias (CSF) para cada línea de células sanguíneas. Los CSF son proteínas difusibles que estimulan la proliferación de las células madre hematopoyéticas. Algunos CSF actúan sobre varios tipos de células madre, mientras que otros sólo estimulan a un tipo. Aunque todavía falta mucho por conocer sobre los puntos de origen y los mecanismos de acción de los CSF, muchos parecen ser producidos de forma local por las células del estroma de la médula ósea y algunos pueden almacenarse en la matriz extracelular local. Los CSF se unen a las células madre diana por un pequeño número de receptores de la superficie. A nivel funcional, los CSF representan mecanismos para estimular la expansión de determinados tipos de células sanguíneas si surge la necesidad. El reconocimiento de la existencia de CSF ha suscitado un notable interés sobre su aplicación clínica en los trastornos caracterizados por una deficiencia de leucocitos (leucopenia).

Eritropoyesis
La eritropoyesis se produce en tres oleadas durante el desarrollo embrionario. La primera oleada comienza con los precursores en el interior del saco vitelino, que producen eritrocitos nucleados primitivos que maduran en el interior del torrente sanguíneo. La segunda oleada también comienza en el interior del saco vitelino, pero las células precursoras colonizan el hígado embrionario y producen la primera generación de eritrocitos fetales definitivos, que serán dominantes durante el período fetal. La tercera oleada se compone de células precursoras que entran en el hígado a partir del mesodermo de la región AGM y de la placenta. Algunas de estas células progenitoras eritroides definitivas envían su progenie directamente desde el hígado al torrente sanguíneo como eritrocitos fetales definitivos. En otros casos serán la semilla de la médula ósea y producirán eritrocitos de tipo adulto en el período fetal tardío.
El linaje de los eritrocitos representa una línea de descendientes de las células CFU-S. Aunque las células progenitoras eritroides están restringidas a formar eritrocitos, existen muchas generaciones de células precursoras. Los primeros estadios de la eritropoyesis se reconocen por el comportamiento de las células precursoras en cultivo, más que por diferencias morfológicas o bioquímicas. Éstas son las denominadas unidades formadoras de brotes eritroides (BFU-E) y CFU eritroides (CFU-E), respondiendo cada una de ellas a estímulos distintos. Los precursores CFU-S pluripotenciales responden a la interleucina 3, un producto de los macrófagos de la médula ósea adulta. Una hormona denominada promotora de la actividad de brote estimula las mitosis en los precursores BFU-E. Una célula CFU-E, que tiene menor capacidad proliferativa que una BFU-E, necesita la presencia de eritropoyetina como factor estimulante.
La eritropoyetina es una glucoproteína que estimula la síntesis del ARN de la globina y se produce por vez primera en el hígado fetal. Posteriormente en el desarrollo, la síntesis se desplaza al riñón, que permanece como punto de producción de eritropoyetina en el adulto. En condiciones de hipoxia (p. ej., pérdida de sangre o gran altitud), la producción de eritropoyetina por los riñones se incrementa, lo que estimula la formación de más hematíes para compensar así el aumento de la demanda. En la eritropoyesis adulta el estadio de CFU-E parece uno de los que mejor responde a estímulos ambientales. La placenta parece ser impermeable a la eritropoyetina y esta propiedad aísla al embrión de los cambios en los niveles maternos de esta sustancia y elimina la influencia de la eritropoyetina fetal sobre el aparato de formación sanguínea de la madre.
Una o dos generaciones después del estadio de CFU-E se pueden reconocer sucesivas generaciones de precursores de eritrocitos por su morfología. El primer estadio reconocible es el proeritroblasto, una célula grande y muy basófila que todavía no produce suficiente hemoglobina como para detectarla mediante análisis citoquímicos. Esta célula tiene un nucléolo grande, mucha cromatina nuclear no condensada, numerosos ribosomas y una elevada concentración de ARNm para la globina. Estas características citológicas son típicas de una célula indiferenciada.
Los siguientes estadios de diferenciación eritroide (basófilos, policromatófilos y eritroblastos ortocromáticos) se caracterizan por cambios progresivos en el balance entre la acumulación de hemoglobina recién sintetizada y la disminución de la maquinaria para la producción de ARN y más tarde del aparato para la síntesis de proteínas. El tamaño global de la célula disminuye y el núcleo se hace cada vez más picnótico (menor y con cromatina más condensada), hasta que al final es expulsado en el estadio de eritrocito ortocromático. Tras la pérdida del núcleo y de la mayor parte de las organelas citoplasmáticas, las células rojas inmaduras, que aún contienen un pequeño número de polisomas, son los reticulocitos. Éstos se liberan a la corriente sanguínea, donde siguen produciendo pequeñas cantidades de hemoglobina durante 1 o 2 días (Figura 3).
El estadio final de la hematopoyesis es el eritrocito maduro, que representa la célula terminal diferenciada, porque ha perdido su núcleo y la mayor parte de sus organelas citoplasmáticas. Los eritrocitos en los embriones son más grandes que sus equivalentes adultos y su vida media es más corta (de 50 a 70 días en el feto frente a 120 días en los adultos).

Síntesis de la hemoglobina y su control
Tanto los eritrocitos como la hemoglobina que contienen pasan por transiciones en sus isoformas durante el desarrollo embrionario. La molécula de hemoglobina adulta es un complejo compuesto de hemo y de cuatro cadenas de globina: dos cadenas alfa y dos beta. Tanto las subunidades alfa como las beta son productos de genes localizados en los cromosomas 16 y 11, respectivamente. Las distintas isoformas de las subunidades se codifican de forma lineal en estos cromosomas.
Durante el período de la hematopoyesis en el saco vitelino, se producen isoformas de globina embrionaria. La primera hemoglobina embrionaria, a veces denominada Gower 1, se compone de dos cadenas ζ (de tipo α) y dos ε (de tipo β). Tras pasar por dos formas de transición, la síntesis de hemoglobina entra en un estadio fetal hacia las 12 semanas, que corresponde con el desplazamiento del lugar de la eritropoyesis desde el saco vitelino al hígado. La hemoglobina fetal está constituida por dos cadenas α de tipo adulto, que se forman muy pronto en la embriogénesis, y dos cadenas γ, la principal isoforma fetal de las cadenas β. La hemoglobina fetal es la forma predominante durante el resto del embarazo. El principal valor adaptativo de esta isoforma fetal de la hemoglobina es que muestra una mayor afinidad por el oxígeno que la forma adulta. Esto es ventajoso para el feto, que depende de las concentraciones maternas de oxígeno. Hacia la semana 30 de la gestación empieza a producirse un cambio gradual de la hemoglobina fetal por la tipo adulta, siendo el tipo 22 el predominante. Existe una variante menor, aunque parecida a nivel funcional, es la 22 (Figura 4).

ANGIOGÉNESIS Y VASCULOGÉNESIS
TIPOS CELULARES EN EL CORAZÓN Y SUS ORÍGENES

El corazón maduro tiene tres capas de células: el endocardio, el epitelio que recubre la capa de una célula de espesor; el miocardio, una red orientada precisamente de los miocitos que realizan el trabajo contráctil del corazón; y el epicardio, un epitelio que cubre la superficie externa del corazón. Más allá de esto, el corazón reside dentro de un saco pericárdico, similar en composición y función a la pleura, que cubre los pulmones.
Gran parte del corazón tiene su origen embrionario de un campo de células mesodérmicas que se refiere como el mesodermo cardiogénico. Las formas primitivas del tubo de corazón como una estructura de dos capas, con un endocardio y el miocardio, ambos tipos de células procedentes de mesodermo cardíaco. El endocardio permanece como un epitelio, pero un subconjunto de células delimitan e invaden la matriz proteínica entre el endocardio y el miocardio para formar las almohadillas endocárdicas. Estas células serán vitales para el desarrollo adecuado de la válvula y completar septación de las aurículas y los ventrículos.
Hay un campo secundario apreciado recientemente de mesodermo cardíaco que entra en el corazón después de la formación del tubo cardíaco primitivo y contribuye principalmente a los ventrículos y la región de flujo de salida del corazón. También denominado el campo del corazón anterior; esta es una población de mesodermo cardíaco anterior y medial al campo corazón original, que rodea el saco aórtico.
Aunque la mayor parte del corazón viene de mesodermo cardíaco, hay algunas poblaciones críticas de células que migran hacia el corazón en desarrollo desde fuera. Éstas son células de la cresta neural, que migran desde los arcos branquiales en desarrollo e inervan el corazón, así como forman la mayor parte del músculo liso de la aorta proximal. La migración de la cresta neural es importante para el sistema de desarrollo de arco aórtico, las arterias pulmonares, y ductus arterioso. Otra población externa de las formas celulares que hacen importantes contribuciones al corazón maduro es un grupo de células que forma dorsal e inferior al tubo cardíaco conocido como el órgano proepicárdico. El origen de estas células es un tema de debate; una teoría más aceptada es que se derivan de primordio hepático. Estas células se expanden como una lámina epitelial que cubre la superficie del corazón para formar el epicardio. Por lo tanto, además de los cardiomiocitos, el corazón está compuesto por las células del epitelio y los orígenes de la cresta neural que migran con precisión espacial y temporal durante el desarrollo cardíaco.
FORMACIÓN DE LA MEDIA LUNA EMBRIONARIO CARDIACA Y TUBO DE CORAZÓN

En sus primeras etapas, existe el embrión como un disco de doble capa de dos láminas epiteliales de células suspendidas entre dos cavidades llenas llenas de líquido, el saco vitelino y la cavidad amniótica. La capa ventral (hacia el saco vitelino) es el hipoblasto, que eventualmente será relegado a estructuras extraembrionarias. La capa dorsal (frente a la cavidad amniótica) es el epiblasto, que en realidad va a formar los 3 capas germinales embrionarias: ectodermo (sistema nervioso y la piel), el mesodermo (corazón, el esqueleto, los músculos y el tejido conectivo), y el endodermo (intestino). Mesodermo y endodermo embrionario se separan del ectodermo primitivo por un proceso conocido como la gastrulación. La gastrulación comienza como una forma de ranura en el epiblasto empezando por el extremo de la cola (caudal) y que se extiende poco a poco cranealmente a un punto final conocido como la placa precordal. La ranura se conoce como la línea primitiva, y su borde de ataque del nodo primitivo. A lo largo de la línea primitiva, las células del epiblasto de la lámina epitelial invaden el espacio potencial entre el epiblasto e hipoblasto. El corazón se forma como una media luna de células craneales a la placa precordal y que se extienden a lo largo de ambos lados. Las células a cada lado de esta región han migrado a través de la porción media de la racha y son precursores de los miocitos ventriculares. Finalmente, las células más laterales y caudal son los que han migrado a través de la parte más posterior de la racha y se convertirá en miocitos auriculares (Figura 6).
LOOPING Y LATERALIDAD DEL TUBO DEL CORAZÓN
La media luna cardíaca es una estructura simétrica y se fusiona en un tubo cardíaco inicialmente simétrica de apariencia. Esto ocurre cuando el ectodermo dorsal a la media luna es plegable para crear el canal neural, y el endodermo inicialmente ventral de la media luna es plegable para crear el intestino anterior. Como las formas del intestino anterior, se mueve dorsalmente, empujando la media luna cardiaca ventralmente y medialmente, de tal manera que los dos brazos de la media luna se fusionan en un tubo directamente en frente del intestino anterior. Como se produce la fusión de los campos del corazón, el corazón ya late. En un feto humano esto ocurre en aproximadamente a los 22 días de gestación. Durante la próxima semana, hay un enorme crecimiento de la cabeza, de modo que el corazón pasa de ser la estructura más "craneal" en el embrión.
A medida que la cabeza crece hacia arriba y sobre el tubo de corazón, el tubo propio corazón comienza a sufrir un cambio drástico en la forma conocida como bucle. Aquí el tubo corazón pierde su simetría. La asimetría izquierda-derecha básica del embrión se establece durante la gastrulación por gradientes de concentración de los factores de Sonic Hedgehog (Shh) y factor de crecimiento fibroblástico 8 (FGF-8). Este gradiente se crea por el movimiento ciliar en el nodo primitivo y provoca una cascada de genes aguas abajo para ser activado, incluyendo nodal, zurdo, y Pitx-2. Morfológicamente, la torsión cardíaca implica el crecimiento diferencial, con una mayor proliferación de miocitos lo largo de la curvatura exterior que interior incluidos los que se añade desde el campo cardíaco secundario. Todo esto sirve para alargar la puerta del tracto de salida y ampliar los ventrículos relativos a las aurículas. Además, hay fuerzas mecánicas de tracción, torsión, y realineación de estructuras del tubo cardíaco primitivo. Se cree que esto implicará el citoesqueleto, incluyendo la miosina no muscular, los microtúbulos, y los haces de actina no musculares.
Looping normal ocurre a la derecha (D-bucle), es decir, el seno venoso y aurículas se mueven hacia la izquierda y posterior, y los ventrículos y bulbo cardíaco se mueven a la derecha y anterior. Es importante destacar que el polo venoso craneal también se mueve de tal manera que se alinee con el polo arterial a lo largo del eje del cuerpo horizontal. El bulbo cardíaco permanece a la derecha del ventrículo primitivo. La curvatura exterior anterior es un área de rápido crecimiento del miocardio y la expansión para formar las cámaras ventriculares. La curvatura interior posterior experimenta un crecimiento más lento y actúa más bien como un punto de apoyo para el proceso de bucle. La curvatura interior del miocardio contribuye al canal atrioventricular y es crítica para la formación de las almohadillas endocárdicas.
ANOMALÍAS EN TORSIÓN CARDIACA
A veces, de enlace puede ocurrir a la izquierda, con el polo venoso y moviendo hacia la derecha aurículas y los ventrículos. Esto se conoce como L-looping y más comúnmente se produce como una entidad que se denomina transposición congénitamente corregida. Cuando la aorta es a la izquierda de la arteria pulmonar (paralelo, no cruzado), la circulación resultante es del ventrículo izquierdo a la arteria pulmonar y del ventrículo derecho a la aorta. Además, a menudo hay otros defectos cardiacos que van desde un defecto septal ventricular con estenosis de la válvula pulmonar a los defectos más complejos que involucran hipoplasia de uno de los ventrículos o hipoplasia, aplasia o malformación de una o más válvulas.
L-bucle también puede estar asociada con defectos más globales conocidos como síndromes de heterotaxia. En heterotaxia, asignaciones de izquierda-derecha a lo largo del eje del cuerpo son aleatorios. Por lo tanto, es posible tener asimetría imagen especular, o simetría bilateral, ya sea con dos lados derecho o dos lados izquierdo. Esto a menudo se denomina isomería derecha o izquierda. Esto puede afectar el abdomen, los pulmones y el corazón. En el abdomen, el intestino, el hígado y el bazo pueden invertirse de normal (situs inversus abdominal). Puede haber mala rotación del intestino y anomalías del árbol biliar. El bazo, siendo una estructura normalmente del lado izquierdo, se puede duplicar en isomerismo izquierdo (poliesplenia), y ausente en isomerización derecha (asplenia). Los pacientes asplénicos requieren profilaxis antibiótica en la infancia para proteger contra las infecciones bacterianas. Los pacientes poliesplénicos tienen isomería derecha de los pulmones y el corazón también. Anormalidades pulmonares en heterotaxia se manifiestan en la anatomía bronquial y pulmonar. El pulmón normal izquierdo está bilobulado y el bronquio principal izquierdo se desplaza por debajo de la arteria pulmonar izquierda (hiparterial). El pulmón derecho normal es trilobulado y el bronquio principal se desplaza por encima de la arteria pulmonar derecha (eparterial). Por lo tanto, el examen cuidadoso de una radiografía de tórax en isomerismo izquierdo revela bronquios hiparterial bilateral y en isomerismo derecho muestra bronquios eparterial bilateral. En el corazón, la unilateralidad de la aurícula es aleatorio en heterotaxia. Aquí las aurículas puede ser normal (situs solitus), derecha izquierda invertida (situs inversus), o poco clara (heterotaxia). Las aurículas se definen prácticamente por su retorno venoso, sistémica a la aurícula derecha y pulmonar hacia la izquierda. Pero en heterotaxia esto no siempre es posible, como las venas pulmonares sistémicas o sistémicas pueden volver a ambas aurículas. Las venas pulmonares que crecen desde los pulmones en desarrollo normalmente se unen a la pared posterior de la aurícula izquierda. Por último, ciertos patrones de defectos cardiacos adicionales a menudo acompañan asplenia / isomerismo derecho. Estos incluyen el fracaso de la tabicación de los ventrículos que resulta en un ventrículo único, el subdesarrollo pulmonar (estenosis o atresia de la válvula pulmonar), la vena cava superior bilateral, y el retorno venoso pulmonar anómalo. Para poliesplenia / isomerismo izquierdo, defectos comúnmente asociados incluyen defectos de cojín endocárdicos, lesiones obstructivas izquierdas como la coartación de la aorta, y la interrupción de la vena cava inferior (VCI) con la continuación ácigos a la vena cava superior.
TABICACIÓN DE LA ENTRADA VENTRICULAR
Cojinetes endocárdicos
Después de que ocurre la torsión cardíaca, la orientación del tubo de corazón ha cambiado drásticamente, pero la progresión del flujo de sangre a través de ella sigue siendo esencialmente de la misma secuencia. Segmentos de entrada y auriculares son hacia la izquierda, y el ventrículo y la salida son a su derecha.
Durante la siguiente fase de desarrollo del corazón, las estructuras auriculoventricular (entrada) y ventriculoarteriales (salida) se tabicarán de manera que hay una válvula de entrada y una salida con válvula para cada ventrículo. En los embriones humanos, esto ocurre durante la semana 7 de la gestación. Estas válvulas se forman principalmente a partir de las almohadillas endocárdicas. Los cojines endocárdicos también son fundamentales para completar el desarrollo ventricular, completando las porciones del tabique adyacentes a las válvulas AV.
Dentro del tubo cardíaco primitivo, incluso antes del bucle, hay constricciones débiles en el surco auriculoventricular y en la formación de conotruncus. Los cojines endocárdicos aparecen inicialmente como hinchazones en el tabique auriculoventricular y segmentos conotruncales del corazón primitivo, a las seis semanas de gestación. Las hinchazones son causadas como células del revestimiento interno del corazón (endocardio) y migran a la matriz extracelular en entre el endocardio y el miocardio. La señalización que implican la familia TGF-β entre el miocardio y el endocardio desempeñan un papel crucial en la iniciación y el mantenimiento de este proceso. Las células de laminación cambian de fenotipo en un proceso conocido como transformación mesenquimal-epitelial.
Tanto en el canal AV y conotruncus, se forman cuatro cojines distintos, nombrados por sus localizaciones anatómicas. Para el canal AV estos son superior, inferior, lateral izquierdo y lateral derecho. En el conotruncus tienen razón, dorsal superior, derecha, izquierda inferior y ventral izquierdo. Los amortiguadores AV y conotruncales están separados con una excepción importante: El cojín conal ventral izquierdo y el cojín AV superiores están en la continuidad a lo largo de la curvatura interna del corazón. Esta proximidad persistirá en el corazón totalmente septado como la continuidad de la válvula aórtica mitral. Los septos auriculoventricular así como los cojines superiores e inferiores crecen y se extienden, finalmente en la fusión en la línea media. Esto da lugar a la separación completa del canal AV en los lados izquierdo (mitral) y derecha (tricúspide). Las válvulas AV forman a partir de restos de los cojines: una válvula mitral hacia la izquierda con dos valvas, y una válvula tricúspide hacia la derecha con tres valvas.

El fracaso de la fusión completa entre los cojines superiores e inferiores en los resultados de la línea media es una hendidura dentro de la valva anterior de la válvula mitral. Esta es una forma leve de un defecto de relieve endocárdico, a menudo asociada con un defecto septal atrial primum y conocido como un canal auriculoventricular parcial. La valva septal de la válvula tricúspide también se forma por estos cojines superiores e inferiores y puede ser anormal, así, aunque esto es menos a menudo clínicamente significativa.
TABICACIÓN VENTRICULAR DEL TRACTO DE SALIDA:

Mientras que los cojines AV forman un quid tridimensional del corazón, los cojines conotruncales forman una espiral tridimensional, completando una rotación de casi 180 grados. Durante la tabicación de los tractos de salida, el bulbo cardíaco primitivo es separable en dos segmentos, el tronco adyacente a la salida de la aorta, y los conos adyacentes al miocardio ventricular. Los cojines inferiores superiores e izquierda son correctas dentro de la región del tronco y contribuirán al tabique aorticopulmonario. Los cojines dorsal derecha y ventral izquierda están dentro de la región de cono y son importantes para la formación de la válvula pulmonar y aórtica, y para la finalización de la tabicación entre los ventrículos a nivel de las válvulas pulmonar y aórtica.
Una vez más, para que este proceso se produzca correctamente, la continuidad de la almohadilla AV superior y la izquierda cojín cono ventral es crítica. Para alinear las grandes arterias septadas directamente en los ventrículos, el tronco arterial debe desplazarse hacia la izquierda. Hay pruebas de modelos de ratones que de no lograr la continuidad de la almohadilla AV superior y dejados resultados cojín cono ventrales en la mala alineación de los dos grandes arterias más el ventrículo derecho, junto con un defecto septal ventricular. Clínicamente, se trata de una entidad conocida como ventrículo derecho de doble salida (DSVD). Parte de la definición clínica de un DSVD es la presencia de un segmento de cono de ambos ventrículos (cono bilateral). Anatómicamente, el cono normal es un "cuello" musculoso dentro del ventrículo derecho entre las válvulas tricúspide y pulmonar. Embriológicamente, esto es un remanente del cono del tubo cardíaco primitivo que es retenida por el ventrículo derecho.
Tracto de salida ventricular
El tracto de salida en desarrollo tiene la complejidad añadida de contribuciones de fuera del tubo cardíaco primitivo. Estos incluyen dos importantes poblaciones de células de la cresta neural, y las células del corazón de campo anterior. Los datos recientes con respecto a la expresión de genes en el campo del corazón secundaria han demostrado papeles recién apreciadas por varios genes ya estudiados en el campo del corazón primaria, incluyendo Hand1, Nkx2-5, Gata4 y MEF2. Defectos de expresión génica aquí pueden provocar anomalías como hipoplasia y del tracto de salida ventriculares derechas, incluyendo la tetralogía de Fallot. La segunda población de células que migran en los tractos de salida en desarrollo es la cresta neural cardiaca. Las células neurales se originan en el rombencéfalo anterior, y migran como una hoja a través de la región faríngea y en los arcos aórticos. El tercer, cuarto y sexto arcos son los sitios principales de la migración de la cresta neural. Las células de la cresta neural rodean los epitelios de las arterias del arco y se extienden en el tronco y el cono proximal. Estas células de la cresta neural también son importantes para el desarrollo de la paratiroides cerca, tiroides y glándulas del timo.
SEPARACIÓN DE AORTA Y LA ARTERIA PULMONAR: DEXTROPOSICIÓN NORMAL
Dentro del segmento de tronco, la tabicación también ocurre durante la remodelación del cono. Esto normalmente se produce como los cojines del tronco fusible en forma de espiral. Comenzando en el nivel de las válvulas AV, la arteria pulmonar de nueva formación es directamente anterior a la aorta. Además a lo largo del tracto de salida, la arteria pulmonar y la aorta son más de izquierda a derecha el uno al otro, ya que la espiral se extiende. Con los arcos de la aorta a la izquierda, el arco transversal proximal pasa por delante de la arteria pulmonar, completando un giro de 180 grados de la espiral. Si este giro de 180 grados no se produce o es incompleta, el resultado es transposición grandes arterias. Esto se define por la válvula aórtica ser anterior a la válvula pulmonar.


FORMACIÓN DE LA VÁLVULA CARDIACA
El desarrollo de la válvula cardiaca comienza con los cojines endocárdicos durante la semana 7 de gestación, pero la formación de atrioventricular maduro (mitral y tricúspide) y las válvulas semilunares (aórtica y pulmonar) toma varias semanas para la terminación. Como los cojines de marcación endocárdica se agrandan, sobresalen en el lumen del corazón. Aquí, a reserva de flujo constante de sangre, las protuberancias se condensan y se alargan.
Las válvulas se someten a una amplia remodelación de la matriz extracelular; en el caso de las válvulas semilunares, esto se traduce en una capa fibrosa (principalmente colágeno) en el lado arterial, una capa esponjosa (glicosaminoglicanos) en el medio, y una capa elástica (elastina) en el lado ventricular. En el caso de las válvulas semilunares, este proceso de remodelación de los cojines endocárdicos continúa a través del último trimestre del embarazo y en el periodo neonatal.

DESARROLLO DE LOS VENTRÍCULOS Y SEPTO
El tabique ventricular es muscular y compuesto por un saliente de miocardio ventricular que se inicia en el vértice del ventrículo primitivo y se extiende dentro de la cavidad ventricular. Como el tabique se extiende hacia arriba, la separación se vuelve casi completa. Existe controversia en cuanto a cómo el tabique se extiende tan rápidamente para formar una pared entre los ventrículos izquierdo y derecho. Parte de la respuesta parece ser la rápida proliferación de estos miocitos, que retienen la capacidad de dividirse incluso como un miocardio de trabajo. Esta propiedad se pierde poco después del nacimiento, por lo que el miocardio maduro es en su mayor parte incapaz de proliferación. Pequeños defectos musculares ventriculares septales (VSD) son muy comunes en los recién nacidos.
Hemos dicho que a medida que el septo muscular, crece y se extiende desde el ápex del ventrículo, la separación entre los ventrículos izquierdo y derecho se convierte en casi completa. Hay dos regiones importantes de todo el tabique ventricular que se completa con cojines endocárdicos. En primer lugar, los cojines endocárdicos AV forman el tabique posterior adyacente a las válvulas AV. En segundo lugar, los cojines conales forman el tabique conal debajo de las grandes arterias.
Los ventrículos izquierdo y derecho se diferencian no sólo por su posición relativa entre sí, sino también en su estructura básica muscular y en su función. Cada ventrículo funcionalmente puede subdividirse en regiones de entrada y salida. El VD, como hemos discutido, tiene un segmento conal por lo tanto se conoce como un ventrículo tripartita. En el corazón maduro, el VI es un sistema de alta presión, conectado a presiones sistémicas, mientras que el VD está conectada a la circulación pulmonar de inferior presión. En el embrión, el miocardio se espesa y se arregla en los procesos conocidos como trabeculación y compactación. Trabeculación se refiere a las proyecciones de tejido muscular, en el lumen del ventrículo, de tal manera que la superficie interna ya no es de paredes lisas, sino estriada. La compactación se refiere a la alineación de los miocitos al azar y sin apretar, como lo son en el miocardio inmaduro, en haces de miocitos apretadas y bien coordinados que trabajan como una unidad en el miocardio maduro.
DESARROLLO DEL ATRIO Y FIBRILACIÓN DEL SEPTUM
Al igual que el tabique ventricular, el tabique auricular comienza como una cresta de tejido muscular, superior y línea media, que se expande para dividir el lado derecho de la aurícula que contiene el orificio del seno venoso desde el lado izquierdo del atrio que contiene el orificio de la común vena pulmonar. Esta estructura se conoce como el septum primum, y crece inferiormente hacia los cojines de fusión AV. Los cojines de fusión de AV se forman la parte inferior del tabique auricular. El septum secundum es mucho más rígido que el septum primum, que bate abierta para permitir de derecha a izquierda el flujo. Después del nacimiento y la separación de la placenta de baja resistencia, la presión de la aurícula izquierda se eleva significativamente y la derivación auricular invierte. Esto hace que el septum primum efectúe un sello contra el tabique secundum, a menudo dejando una pequeña abertura conocido como el foramen oval. Un foramen oval permeable, por lo general de 3 mm o menos con un grado trivial de flujo de izquierda a derecha, es un hallazgo normal en un recién nacido e incluso en muchos niños mayores y adultos. Un defecto mayor de 5 mm puede constituir un defecto septal auricular secundum (CIA), una verdadera deficiencia en el tejido septal auricular en la región del foramen oval.

DESARROLLO VENOSO SISTÉMICO Y PULMONAR

El seno venoso del tubo cardíaco primitivo es una estructura simétrica y recibe tres conjuntos de pares de venas sistémicas (izquierdo y derecho): el vitelino, umbilical y cardinales comunes. Esta es la situación en la gestación de 4 a 5 semanas.
Las venas vitelinas regresan del saco vitelino, una estructura que se comunica con el intestino primitivo a través del conducto de línea vitelina.
Las venas umbilicales regresan de la placenta trayendo sangre oxigenada. Estos son los primeros en desarrollar, por la gestación de 3 semanas. A medida que se desarrolla el hígado, las venas umbilicales desarrollan conexiones con el plexo venoso del hígado, y la conexión los involuciona a un seno venoso. Fuera del embrión y dentro del cordón umbilical, las venas umbilicales izquierdo y derecho se funden en uno; por lo tanto hay una sola vena umbilical en el recién nacido. Dentro del embrión, los segmentos izquierdo y derecho se mantienen separados, y la vena umbilical izquierda se convierte conectados a las venas hepáticas derecha a través de un nuevo canal que forma, el conducto venoso. Esta circulación está formado por la séptima semana de gestación. Una vez más las venas umbilicales están llevando sangre oxigenada a la aurícula derecha. Este flujo se dirige principalmente hacia el septo auricular, y por lo tanto principalmente a través del foramen oval en la aurícula izquierda y la circulación sistémica.
Las venas cardinales llevan el retorno venoso desde el embrión propiamente dicho. Las venas cardinales comunes izquierda y derecha son segmentos relativamente cortos que se conectan al seno venoso. A cada lado, anterior y venas cardinales posteriores se unen para formar la vena cardinal común; el cardinal anterior se encarga del retorno venoso de la parte superior del cuerpo y el posterior de la parte inferior del cuerpo. La vena cardinal común derecha será la vena cava superior en la circulación maduro. La vena cardinal común izquierda y el segmento izquierdo del seno venoso forman el seno coronario, que recibe el retorno venoso desde el sistema coronario.
Las venas pulmonares crecen en progresivamente a partir de la vasculatura pulmonar en desarrollo, primero como una vena pulmonar común de ambos pulmones. Esta vena se funde en la parte posterior de la aurícula izquierda. A medida que el atrio se expande, la vena pulmonar común se absorbe cada vez más en la pared trasera, de tal manera que primero 2, a continuación, 4 de sus ramas distales eventualmente entrar independientemente en la aurícula izquierda, 2 desde la izquierda y 2 desde el pulmón derecho. El segmento auricular original del tubo de corazón no es parte de las aurículas que funcionan en el corazón maduro, en vez relegados a la derecha e izquierda apéndices auriculares.
DESARROLLO DEL CAYADO AÓRTICO

El tubo cardíaco primitivo se conecta a la salida de la aorta, el precursor de la aorta ascendente, y comienza a bombear a la circulación sistémica en desarrollo del embrión tan pronto como se forma. El saco aórtico se conecta a dos aortas dorsales posteriormente a través de una raíz aórtica, lo que da lugar a una serie de arcos, los arcos branquiales. Nuevamente, estas estructuras están emparejados, y hay un total de seis pares, aunque no todos son patentes al mismo tiempo. Arcos 1 a 4 llevan el flujo sanguíneo en la 4 semana de gestación.
Arcos 1 y 2 en su mayoría retroceden completamente, aunque algunas partes contribuyen a algunas de las arterias de la cara. Arco 3 no persiste como un arco, pero contribuye a la formación de las arterias carótidas, tanto izquierdo y derecho. La extremidad izquierda de arco 4 permanece en continuidad con la raíz aórtica, juntos haciendo hasta el verdadero arco aórtico y su primera ramificación, la arteria braquiocefálica (innominada). La extremidad derecha del arco 4 se convierte en parte de la arteria subclavia derecha, que conserva su conexión proximal con la arteria braquiocefálica. Esta remodelación arco se ha producido por 7 semanas de gestación. En algunos casos, ya sea de forma independiente o como parte de otro defecto cardíaco, la extremidad derecha del arco de 4 sigue siendo patente y la izquierda constituye la arteria subclavia izquierda. Arco 5 es pequeño y nunca desarrolla plenamente. El arco 6 forma con el sistema vascular de la arteria pulmonar en desarrollo y constituye las arterias pulmonares izquierda y derecha y la arterioso ductus arterioso. El arco ductal persiste durante toda la vida del feto como otro medio de shunt derecha a izquierda, permitiendo que la sangre desde el ventrículo derecho a pasar por alto la circulación pulmonar y cruzar a la aorta descendente.
ARTERIAS CORONARIAS
Las arterias coronarias fueron durante muchos años asumidas brotar de la aorta ascendente y crecer sobre la superficie del corazón. Esto ahora se sabe que es incorrecta. Las arterias coronarias se derivan de la epicardio del corazón. Temprano en el desarrollo, grupos bilaterales de células aparecen cerca del septo transverso, donde el brote de hígado se acerca al corazón. Estas células se extienden sobre la superficie del corazón como un epitelio, formando el epicardio. Los vasos coronarios forman, crecen, y se fusionan uno con el otro para formar una vasculatura de trabajo. Esto corresponde con el proceso de crecimiento y trabeculación de los ventrículos. Con el aumento de la complejidad y el grosor del miocardio, la difusión simple de nutrientes de la sangre dentro del corazón ya no es adecuado. La vasculatura coronaria, a continuación, proporciona el suministro de sangre necesario un miocardio de trabajo mediante la ampliación de sus buques de superficie a la derecha y a la izquierda para conectar a la aorta justo por encima de la válvula aórtica.
RESUMEN DE LA EMBRIOLOGÍA CARDIOVASCULAR
El desarrollo embrionario comienza desde la fecundación y se extiende hasta finales de la semana 8 de gestación, ya que a partir de la semana 9 el feto ya está completamente formado y crece progresivamente durante el periodo restante de la gestación hasta que sobreviene el nacimiento. El sistema cardiovascular en el ser humano comienza a aparecer hacia la mitad de la semana 3 de desarrollo intrauterino.
Al inicio, las sustancias nutritivas se difunden desde la sangre materna al interior de una serie de canalículos, y la circulación de los líquidos por estos canales se realiza por difusión y ósmosis. Posteriormente se diferencia el elemento impulsor principal que dará energía y rapidez a la circulación: el corazón.
DESARROLLO DEL CORAZÓN
Se inicia en la placa cardiógena del mesodermo embrionario con la formación del tubo cardiaco y termina con la formación del corazón definitivo.
Formación del tubo cardiaco
Al final de la tercera semana (19 día) surgen en la hoja esplácnica de la placa cardiógena del mesodermo unas agrupaciones celulares, los islotes wolffianos, que forman dos cordones macizos a cada lado de la línea media en dirección craneocaudal. Se establecen unos puentes entre ambos, que al aumentar forman el esbozo cardiaco único. El esbozo por ahuecamiento constituye el tuvo cardiaco. Para el día 22 se establece este tubo único, de situación media y longitudinal, que representa el revestimiento endotelial del corazón o futuro endocardio.
Constitución del tubo cardiaco
El tubo cardiaco se alarga y desarrolla dilataciones alternadas con estrechamientos. Las primeras dilataciones que aparecen (día 22) son el bulbo arterial, el ventrículo primitivo y la aurícula. El seno venoso se ve un día después (día 23) y es la dilatación más caudal, que consiste en una porción transversal pequeña y prolongaciones derecha e izquierda.
Al formarse el endotelio del tubo cardiaco, el mesénquima que lo rodea se torna grueso y forma la hoja mioepicárdica, que al principio está separada de la pared endotelial por la gelatina cardiaca o jalea de Davis. El tubo endotelial será el endocardio, y la hoja mioepicárdica dará origen al miocardio y al epicardio o pericardio visceral.
Configuración externa del tubo cardiaco
Los cambios en la conformación externa del tubo cardiaco se evidencian en las porciones bulboventricular (que es el segmento arterial) y auriculosinusal (que es el segmento venoso). El tubo cardiaco experimenta una acotadura (días 21 a 22) y prosigue el desarrollo desigual de sus dos componentes. El ventrículo primitivo se desarrolla más que el bulbo arterial, lo que conduce a la formación de una asa en forma de U en la que una de sus ramas toma posición ventral y la otra dorsal. El bulbo arterial forma la rama ventral (origina los infundíbulos de ambos ventrículos y la posición ascendente de la aorta y pulmonar) y el ventrículo primitivo constituye la parte convexa de la U (origina los dos ventrículos, entre los que aparece un surco interventricular que es consecuencia de la formación del tabique interventricular). La porción auriculosinusal en un principio se encuentra fuera de la cavidad pericárdica y posteriormente ingresa en ella, luego experimenta diferentes modificaciones que provocan que en la aurícula izquierda desemboquen las cuatro venas pulmonares, y las orejuelas auriculares derecha e izquierda surgen por el crecimiento de la aurícula que envuelve el bulbo arterial.
Configuración interna del tubo cardiaco
Empieza a desarrollarse una serie de tabiques y repliegues que transforman su cavidad única en cuatro cavidades: las aurículas y ventrículos definitivos. Este proceso de tabicación comienza el día 27 y termina el día 37, cuando el embrión aumenta su longitud de 5 a 17 mm.
Tabicación del conducto auriculoventricular
El tejido subendocárdico de este conducto prolifera en sus caras anterior y posterior, se dirige hacia el centro del conducto y forma las almohadillas endocárdicas. Las almohadillas se fusionan para formar el septum intermedio y crecen también en sentido cefálico y caudal hacia la aurícula y el ventrículo, contribuyendo a la formación de los respectivos tabiques que separan estas cavidades.
Tabicación del bulbo arterial
La finalidad de esta tabicación es dividir al bulbo en dos conductos, que serán los infundíbulos de los ventrículos y las porciones ascendentes de la aorta y de la arteria pulmonar. En la pared interna del bulbo aparecen las llamadas crestas bulbares (fin de la semana 5). Con el crecimiento, las crestas bulbares terminan por juntarse y fusionarse por su borde libre para formar el tabique aorticopulmonar. Así, queda tabicado el bulbo y constituido el origen de las arterias aorta y pulmonar, como dos vasos enrollados en espiral. La orientación definitiva de ambos vasos en el tórax se debe a la posición que adopta el corazón (ápex hacia adelante, a la izquierda y hacia abajo).
Tabicación interventricular
Aparece una cresta en la mitad de las caras anterior, inferior y posterior del ventrículo, originada por un crecimiento del miocardio hacia el septum intermedio y el tabique aorticopulmonar. Esta cresta, que está situada al inicio del tabique, es una gruesa lámina dispuesta en plano sagital y con su borde libre semicircular, y se conoce con el nombre de septum interventricular primitivo. Las dos crestas bulbares proliferan y cierran la mitad anterior y ventral del foramen, y la almohadilla endocárdica posterior crece para cerrar la parte posterior del foramen.
Tabique interauricular
Los fenómenos que intervienen en la zona que comprende el seno venoso y la aurícula común para formar el tabique interauricular siguen una cronología en su formación morfológica.
Septum primum: Es la membrana delgada y falciforme que aparece en la pared posterior, superior y anterior de la cavidad auricular primitiva y crece hacia las almohadillas endocárdicas.
Osteum primum: Los dos extremos del septum primum toman contacto con las almohadillas endocárdicas. La parte central del borde libre inferior del septum primum y el septum intermedio constituye una abertura temporal grande, el ostium primum, que comunica las aurículas primitivas derecha e izquierda.
Osteum secundum: El ostium primum se va cerrando por proliferación de la almohadilla endocárdica anterior, pero antes de que culmine la obliteración (inicio de la semana 7) aparecen en la parte superior del septum primum una serie de perforaciones que se unen para formar otro orificio, el ostium secundum.
Septum secundum: Al final de la semana 7 aparece a la derecha del septum primum una nueva membrana semilunar, el septum secundum, que cierra el ostium secundum sin adherirse a él, a medida que avanza hacia la pared posterior de la aurícula.
Agujero oval y conducto de Botal: La concavidad entre los dos cuernos del septum secundum se conoce como agujero o foramen oval, y la hendidura que comunica a una aurícula con otra, a través de este orificio y el ostium secundum, se denomina conducto de Botal.
El el momento del nacimiento, al aumentar la presión en la aurícula izquierda, el septum primum se desvía y se pone en contacto con el septum secundum para cerrar el agujero oval. Por ello, el septum primum también se conoce como válvula del agujero oval.
Formación de las válvulas cardiacas
En el interior de los orificios auriculoventriculares y del bulbo arterial aparecen unos engrosamientos que se transforman en las válvulas auriculoventriculares (tricúspide y mitral) y ventriculoarteriales (sigmoideas aórtica y pulmonar).
Formación del sistema de conducción
Deriva del mismo mesénquima cardiogénico. El nódulo sinoauricular se desarrolla en la parte correspondiente al seno venoso y más tarde queda situad en la pared de la aurícula derecha, junto a la desembocadura de la vena cava superior. El nódulo auriculoventricular tiene un doble origen, derivado en parte de la musculatura de la prolongación izquierda del seno venoso y en parte de la musculatura del canal auriculoventricular.
La cardiogénesis ocurre en un intervalo de 3 días a partir del día 23 y a los 2.5 mm de longitud del embrión. El corazón empieza a latir según la secuencia en sentido cefalocaudal. La primera zona en la que aparecen contracciones es la región del bulbo arterial y luego aparecen en el ventrículo, la aurícula y el seno venoso.
DESARROLLO DEL SISTEMA VASCULAR
Simultáneamente aparecen en distintas partes del mesodermo acumulaciones celulares conocidas como islotes de Wolff y Pander, que experimentan una serie de transformaciones. Las acumulaciones celulares forman cordones macizos, que se convierten en tubos cuando aparecen espacios entre sus células. Las células periféricas de estos tubos se aplanan y forman el endotelio de los vasos, y las células centrales se convierten en células sanguíneas primitivas. Las capas de músculo liso y tejido conectivo de los tubos se desarrollan a partir del mesénquima que los rodea.
Sistema arterial
El sistema arterial está representado por las aortas primitivas, cuyas colaterales son los arcos aórticos y las arterias segmentarias ventrales (vitelinas y umbilicales), laterales y dorsales.
Aortas primitivas
En épocas tempranas las aortas primitivas son dos vasos longitudinales que se confunden en el extremo cefálico del tubo cardiaco en uno solo: el saco aórtico, que se continúa en el tronco arterioso del tubo cardiaco. Posteriormente, las aortas se fusionan en la 7ª somita cervical en dirección caudal y forman la aorta descendente definitiva, cuya porción final es la sacra media. Los dos troncos aórticos desprenden colaterales que se distribuyen por todo el embrión. Para la región cefálica originan los arcos aórticos y para el resto del embrión dan origen a las arterias segmentarias ventrales, laterales y dorsales.
Arcos aórticos
Originan las arterias de la cabeza y el cuello. Se inician como puentes vasculares tendidos entre el saco aórtico y la porción cefálica descendente de las aortas dorsales. Estos puentes surgen del seno mesenquimatoso de los arcos faríngeos (arcos 1º a 6º), y luego dan origen a los troncos supraaórticos (salvo el 5º arco, que se oblitera sin dejar huella). El 4º arco aórtico embrionario forma el arco aórtico, y el 6º arco izquierdo forma la arteria pulmonar y el conducto arterioso.
Arterias segmentarias ventrales, laterales y dorsales
Las arterias segmentarias ventrales son colaterales que se distribuyen en el intestino primitivo. En la región torácica surgen las arterias bronquiales y esofágicas. En la región abdominal los vasos se prolongan hasta el saco vitelino (arterias vitelinas) y hasta la placenta (arterias umbilicales). De las arterias vitelinas, al atrofiarse el saco vitelino se crean tres sistemas arteriales: tronco celíaco, mesentérica superior y mesentérica inferior, destinadas a los derivados del intestino posterior y la cloaca. La arteria umbilical permanece doble y está representada en el adulto por las ilíacas primitivas, ilíacas internas y vesicales superiores. El resto de cada arteria umbilical está representada por cordones fibrosos a cada lado del uraco, desde la vejiga primaria hasta el ombligo. Por último, generan ramas destinadas, casi todas, a los derivados de las crestas urogenitales, que son las arterias renales, gonadales, suprarrenales y frénicas inferiores.
Sistema venoso
El sistema venoso está representado por el sistema cardinal (drena el embrión), las venas umbilicales (placenta) y las venas vitelinas (saco vitelino y vasos vitelinos). Las modificaciones del sistema venoso incluyen la instalación de un sistema hepatoportal definitivo y de las venas cavas superior e inferior. Las venas umbilicales empiezan a transformarse durante la semana 5 y finalizan alrededor de la semana 6 a medida que ambas venas umbilicales, derecha e izquierda, se dirigen hacia el seno venoso, se ponen en contacto con el hígado en expansión y son perforadas por las sinusoides hepatovitelinas. La sangre que fluye por la vena umbilical se desvía hacia los sinusoides hepatovitelinos y llega por su intermedio a la vena cava inferior. También desaparece el resto de la vena umbilical derecha, pero no la porción distal de la vena umbilical izquierda, que se hipertrofia y hace pasar la sangre que desde la placenta llega al plexo de sinusoides. Uno de los sinusoides se dilata hasta alcanzar un calibre mayor, y comunica la porción hepática de la vena cava inferior con la umbilical izquierda; es el llamado conducto venoso de Arancio y conduce gran parte de la sangre de la placenta a la vena cava inferior, son derivar por el plexo sinusoide. Después del nacimiento, al cesar la circulación placentaria, la vena umbilical izquierda (la única que quedaba) se transforma en el ligamento redondo del hígado, y el conducto venoso de Arancio se transforma en el ligamento venoso.
FACTORES GENÉTICOS IMPLICADOS EN EL DESARROLLO NORMAL Y DE LAS CARDIOPATÍAS CONGÉNITAS
Los métodos moleculares de identificación de linajes celulares, experimentación con modelos transgénicos y análisis clonal retrospectivo son las nuevas estrategias en el estudio de la cardiogénesis. Durante el desarrollo embrionario del sistema cardiovascular, a los 19 días de gestación, las células cardiacas progenitoras especializadas se agrupan y forman estructuras conocidas como áreas cardiogénicas, en las que comienzan a expresarse genes característicos del miocardio como NKX2.5 y GATA4. La expresión del primer gen depende de proteínas secretadas por el endodermo subyacente, como cerberus, la proteína morfogenética ósea (BMP) y el factor de crecimiento de fibroblastos de tipo 8 (FGF-8). A las 3 semanas de desarrollo las células de este primordio cardiaco migran hacia la línea media y forman en el mesodermo esplácnico de la placa cardiógena el tubo cardiaco.

Los genes expresados en la placa cardiogénica, como NKX2.5, el factor de transcripción de unión al factor de respuesta sérica c-fos (SRF), GATA4, TB5 y HAND2 forman el centro regulador de la red de morfogénesis cardiaca, la cual controla la rotación del tubo cardiaco, la simetría izquierda - derecha y la formación de las cámaras cardiacas. Las células del primer campo del corazón (first heart field, FHF) contribuyen únicamente a la formación del ventrículo derecho y del defecto auriculoventricular, mientras que las aurículas, el ventrículo izquierdo y gran parte del tracto de salida provienen de precursores mesenquimatosos que residen en el segundo campo del corazón (second heart field, SHF).
Defectos de la tabicación y de la conexión auriculoventricular
Los defectos de la tabicación representan el 50% de las cardiopatías congénitas. Los trastornos de los factores de transcripción NKX2.5, TBX5 y GATA4, impiden que se desarrolle normalmente el proceso de tabicación auricular y ventricular.
GATA4 es un factor de transcripción que trabaja junto con NKX2.5. Se ha relacionado con defectos interauriculares e interventriculares sin la presencia de defectos de conducción. Se ha relacionado con defectos de doble salida del ventrículo derecho y ventrículo izquierdo hipoplásico.
El síndrome de Holt - Oram es causado por mutaciones en TBX5 y se caracteriza por defectos del tabique auricular y ventricular similares a los observados en mutaciones de NKX2.5 asociados a malformaciones en brazos y piernas.
MYH6 es activado por GATA4 y TBX5, y se ha relacionado con defectos del tabique ventricular. TBX20 interactúa con NKX2.5, GATA4 y TBX5. Las mutaciones en la caja t (t-box) de este gen se asocian con distintas anomalías que incluyen defectos de tabicación, valvulogénesis y miocardiopatía dilatada en adultos.
Defectos del tracto de salida y del arco aórtico
La deleción 22q11 es el tipo de deleción más frecuente y es la segunda causa de cardiopatía asociada a un síndrome, después de la trisomía 21. Se caracteriza por malformaciones asociadas a defectos en los arcos y bolsas faríngeas, como paladar hendido e hipoplasia tímica y paratiroidea, además de una serie de cardiopatías congénitas de las cuales la más común es la persistencia del conducto arterioso, e incluyen la tetralogía de Fallot, la interrupción del arco aórtico y la doble salida ventricular.
En el síndrome de Char, una afección autosómica dominante caracterizada por persistencia del conducto arterioso, facies dismórfica y alteraciones digitales, es causado por alteraciones del factor de transcripción (TFAP-2 beta). Este gen se expresa principalmente en las células de la cresta neural, que tienen un papel importante en la tabicación que se forma entre la aorta y la arteria pulmonar en el tracto primitivo común.
Defectos obstructivos de la arteria pulmonar y de la aorta
Los defectos obstructivos tanto de la arteria aorta como de la pulmonar varían en su intensidad y pueden llegar, en su peor extremo, a la hipoplasia ventricular. El síndrome de Williams se caracteriza, desde el punto de vista cardiovascular, por estenosis supravalvular aórtica y estenosis periférica de las arterias pulmonares. Muestra, además, varias características extravasculares, como retraso mental, hipercalcemia neonatal, etc. La microdeleción en este síndrome, 7q11, conlleva haploinsuficiencia en el gen de la elastina (ELN), causante de anomalías vasculares.
El síndrome de Noonan se caracteriza por estatura corta, anormalidades faciales y anormalidades cardiacas, principalmente estenosis pulmonar asociada con una válvula pulmonar displásica. Se han encontrado mutaciones puntuales con ganancia de función en el gen PTPN11 en cerca del 50% de los pacientes. El mecanismo parece ser hipoproliferación de los cojines del tracto de salida, estructuras de las que derivan las válvulas arteriales.
Un síndrome que incluye en su morfología alteraciones congénitas del corazón derecho es el de Alagille. En él se presentan distintas anomalías, desde estenosis pulmonar moderada hasta tetralogía de Fallot, además de alteraciones extracardiacas como hipoplasia de vías biliares. El fenotipo es causado por mutaciones o deleciones que comprenden el locus del gen JAGGED-1 hasta en el 94% de los casos. También se han identificado mutaciones en JAGGED-1 en pacientes con estenosis pulmonar y tetralogía de Fallot sin otras características del síndrome. JAGGED-1 es un ligando para el grupo NOTCH1-4, una familia de receptores transmembrana relacionados con la diferenciación celular y el desarrollo embrionario (Tabla 1)
CARDIOPATÍA
|
GENES IMPLICADOS
|
Comunicación interventricular
|
NKX2.5, GATA, TBX20, TBX1, TBX5
|
Comunicación interauricular
|
NKX2.5, GATA, TBX20, MYH6, TBX5, GATA6
|
Estenosis pulmonar
|
JAG1, NOTCH2, PTPN11
|
Defecto auriculoventricular
|
TBX5, NKX2.5, CRELD1, PTPN11, KRAS, SOS1, RAF1, GATA6
|
Tetralogía de Fallot
|
NKX2.5, NOTCH1, TBX1, JAG1, NOTCH2, GATA6, TBX20, CITED2
|
Persistencia del conducto arterioso
|
TFAP2B
|
Transposición de grandes vasos
|
NKX2.5, THRAP2
|
Estenosis valvular aórtica
|
NOTCH1, PTP11
|
Coartación aórtica
|
NOTCH1, PTP11
|
Doble salida del ventrículo derecho
|
NKX2.5, THRAP2, GATA4
|
Hipoplasia del ventrículo izquierdo
|
NOTCH1, NKX2.5
|
Tronco arterioso
|
TBX1, GATA6
|
Conexión anómala de venas pulmonares
|
TBX20
|
Atresia tricuspidea
|
NKX2.5
|
Anomalía de Ebstein
|
NKX2.5
|
Modificado de: Moro M., et al (2014). Tratado de Pediatría. Asociación Española de Pediatría. 11º Edición. Médica Panamericana. Argentina.
|
|
EXPLORACIÓN FÍSICA CARDIOCIRCULATORIA
El corazón está situado en la cavidad torácica y funciona como una bomba que impulsa la sangre a todo el cuerpo. Se divide en cuatro cavidades: dos aurículas (derecha e izquierda) que son cámaras de recepción; y dos ventrículos (derecho e izquierdo) que funcionan como cámaras de expulsión.
Un ciclo cardiaco está formado por una fase de relajación (diástole) y una fase de contracción (sístole). El corazón se forma entre la tercera y la séptima semana del periodo embrionario.
En la etapa fetal, la sangre fluye de la placenta materna a la vena umbilical del feto, esta sangre contiene aproximadamente 35 mmHg, pasa a través del conducto venoso, donde drena a la vena cava inferior hasta llegar a la aurícula derecha, pasa por el foramen oval hacia la aurícula izquierda, se mezcla con la escasa sangre que retorna de los pulmones por las venas pulmonares, y de allí es expulsada por el ventrículo izquierdo (VI) hacia la aorta ascendente para proporcionar sangre oxigenada (con una PO2 de aproximadamente 28 mmHg) a las arterias coronarias, cerebro y extremidades superiores. Al llegar a estos puntos, la sangre tiene una PO2 elevada.
La vena cava superior drena sangre desoxigenada del cerebro a la aurícula derecha, para luego pasar al ventrículo derecho (VD). El 90% de esta sangre es eyectada a través del conducto arterioso hacia la aorta, el 10% es expulsado hacia la arteria pulmonar y los pulmones, limitada debido a la resistencia vascular pulmonar. La PO2 en el VD es de 19 a 21 mmHg.
Finalmente, la aorta drena de regreso a la placenta a través de las dos arterias umbilicales mediadas por las arterias iliacas. La PO2 de la sangre que retorna a la placenta es de 20 a 25 mmHg. La placenta tiene baja resistencia vascular, por lo que acepta el retorno de sangre arterial.
Al momento del nacimiento existe un incremento súbito de la poscarga sistémica al excluirse la placenta de baja resistencia. La precarga en el lado derecho disminuye al cesar el retorno venoso umbilical. La resistencia vascular pulmonar se reduce debido a la expansión pulmonar durante la primera respiración y al aumento de presión parcial de oxígeno de los alvéolos, así como la intervención de vasodilatadores pulmonares. Esto disminuye las presiones del VD y una reducción del efecto restrictivo del VD dentro del pericardio. La presión intrapericárdica se reduce con la ventilación espontánea. Estos cambios ayudan a adaptar al VI a la precarga aumentada por el retorno venoso pulmonar.
El corazón derecho del neonato recibe sangre poco oxigenada desde la vena cava inferior que transporta la sangre procedente del tórax, abdomen y extremidades inferiores; así como la vena cava superior que recibe sangre de las extremidades superiores y de la cabeza. Esta sangre llega a la aurícula derecha y pasa al VD a través de la válvula tricúspide, luego hacia los pulmones donde se oxigena por medio de las arterias pulmonares; una vez oxigenada, entra a la aurícula izquierda por las venas pulmonares y pasa al VI por la válvula mitral. La sangre es expulsada hacia la aorta, a través de la válvula aórtica.
La detección de un soplo es un signo de cardiopatía, pero es muy poco específico, dado que está presente en muchos niños sanos (entre el 50 al 90% en algún momento de su vida). Una buena exploración física cardiovascular permitirá al pediatra descartar la existencia de alteraciones con lo que se evitará la realización de exploraciones complementarias innecesarias en la mayoría de los casos.
Antes de comenzar con la exploración física propiamente dicha, es muy importante cuidar las condiciones en que se van a realizar, ya que disponer de un ambiente adecuado hará que ésta se efectúe con mayor éxito. Tras establecer una buena empatía con el paciente y su familia, y tras realizar una anamnesis minuciosa, se debe lograr que el paciente se encuentre en decúbito supino sobre la camilla, con el tórax descubierto y el explorador a la derecha, preferiblemente sentado. Es importante la comodidad de ambos, así como el silencio adecuado. Ésta puede ser alterada en lactantes poco colaboradores, anticipando la auscultación incluso en brazos de los padres, para conseguir el objetivo.
Se obtendrán las constantes vitales básicas: frecuencia cardiaca, respiratoria y presión arterial. Los valores normales varían ampliamente con la edad. Para la determinación de la presión arterial hay que utilizar el manguito adecuado al paciente, esto es, aquel cuya anchura sea al menos el 40% del perímetro del brazo y la bolsa inflable rodee al menos el 80% del perímetro del brazo. La presión debe tomarse en ambas extremidades superiores y al menos en una extremidad inferior. El niño debe permanecer relajado, sentado y con el brazo en reposo a la altura del corazón (Figura 15).

INSPECCIÓN
Permite apreciar de conjunto a nuestro paciente; incluye su relación e interacción con el medio ambiente, su capacidad física para movilizarse y su estado nutricional, entre otros aspectos.
Se inicia con la apreciación del color de la piel del niño; la palidez de esta sugiere la existencia de anemia, vasoconstricción o cianosis. Para que aparezca cianosis verdadera se necesita que la saturación de oxígeno arterial experimente una disminución significativa; sin embargo, la sangre arterial no suele ser visible y el color de la piel depende básicamente del plexo venoso subcutáneo. En circunstancias normales, en los capilares cutáneos se extrae poco oxígeno y la saturación de este gas en la sangre venosa es relativamente alta, salvo que la circulación se identifique por la constricción arteriolar causada por el frío o por un mayor tono adrenérgico. Por las razones expuestas, en el mejor de los casos, la observación permite contar solo con una base indirecta para juzgar cuál es la saturación arterial, y, generalmente, no se reconoce la cianosis clínica hasta que aquélla cae por debajo del 85%.
Es necesario señalar que ante toda sospecha de cardiopatía congénita debe valorarse la presión parcial de oxígeno (PO2) arterial y sus variaciones al respirar el paciente y las diferentes concentraciones de este gas (FiO2). La PO2 de un recién nacido normal al respirar aire oscila entre 60 y 80 mm Hg; cifras menores hacen pensar en la existencia de cortocircuito venoarterial. Para un FiO2 del 100%, la PO2 alcanza valores muy altos en ausencia de cardiopatía congénita (alrededor de los 400 mm Hg) y, contrariamente, las variaciones son mínimas si el paciente presenta malformaciones cardiovasculares. Existen 3 circunstancias en las que en ausencia de cardiopatía congénita resulta escaso el aumento de la PO2 con ascensos en el FiO2 hasta el 100 %: neumópatas con cortocircuito intrapulmonar (atelectasia o neumotórax), fístulas arteriovenosas pulmonares y síndrome de circulación fetal persistente; en este último la anormal persistencia de resistencias pulmonares elevadas origina un cortocircuito venoarterial vía auricular (foramen oval) con cianosis severa como expresión del cortocircuito intracardíaco. Además de la cianosis existen otras características dermatológicas que se deben tener en cuenta en la inspección del aparato cardiovascular como son: sequedad o aspereza de la piel, sudación excesiva, presencia de cicatrices torácicas, etc.
El análisis visual morfológico de la porción distal de los miembros es de gran utilidad; la existencia de dedos hipocráticos (uñas en “vidrio de reloj” y dedos en “palillo de tambor”) nos habla a favor de la presencia de hipoxemia de largo tiempo de evolución; la configuración digital o de las manos puede aproximarnos al diagnóstico de diversas alteraciones metabólicas, genéticas o hereditarias asociadas a malformaciones cardiovasculares como los síndromes de Turner, Down, Rubinstein - Taybi, Holt - Oram y la trisomía 18, entre otros. No debemos olvidar que la presencia de hemorragias “en astilla” del lecho ungueal, de nódulos de Osler o lesiones de Janeway evidencian clínicamente alteraciones inmunoalérgicas propias de la endocarditis infecciosa.
La inspección de la región anterior torácica, que por encontrarse situada delante del corazón y de los grandes vasos recibe el nombre de región precordial, reviste gran interés. El área cardíaca precordial puede ser normal, presentar deformidad hacia la punta, en el borde paraesternal izquierdo o global; ello significa aumento del área cardíaca, que puede ser a expensas del ventrículo izquierdo, del derecho o de ambos. El abombamiento precordial es ex- presión de cardiomegalia crónica a punto de partida de sobrecarga volumétrica como la que ocurre debido a un gran cortocircuito de izquierda a derecha.
El latido apexiano (de la punta o del ventrículo izquierdo) es un movimiento precordial expansivo, de breve duración y de pequeña amplitud, como un choque, que ocurre al principio de la sístole, no se aprecia en la primera infancia a no ser que el ventrículo izquierdo esté dilatado o el tórax carezca del tejido adiposo subcutáneo. Debido a la posición relativamente horizontal del corazón, el latido apexiano se observa, generalmente, en el cuarto espacio intercostal, por fuera de la línea medioclavicular hasta los 3 años de edad; posteriormente se localiza al nivel del quinto espacio intercostal en la línea medioclavicular, ubicado en una pequeña área de 2 a 3 cm de diámetro (la zona de la areola mamaria corresponde al cuarto-quinto espacio intercostal). El latido apexiano se debe al retroceso del corazón cuando expulsa la sangre, aunque se plantea la intervención de otro factor constituido por la ligera rotación del órgano hacia delante y a la derecha, que favorece la proyección de la punta contra la pared del tórax. El latido de la punta puede ser visible, desplazado hacia abajo y afuera o hacia arriba y adentro.
Cuando existe crecimiento masivo del ventrículo derecho se puede observar abombamiento precordial y presencia de latido epigástrico en el recién nacido y lactante pequeño o latido paraesternal bajo en el niño mayor. En caso de crecimiento del ventrículo izquierdo se puede denotar el latido apexiano desplazado hacia abajo y afuera con un carácter reptante o en escalera.
El examinador no debe dejar de observar los espacios intercostales izquierdos segundo y tercero, donde pueden visualizarse latidos en los pacientes con hipertensión pulmonar. Asimismo, es posible localizar latidos en la horquilla esternal en aquellos niños que presentan estenosis o coartación de la aorta y la llamada “danza arterial carotídea cervical” en los casos que padecen insuficiencia aórtica de cierta consideración.
Es necesario señalar que deformidades estructurales torácicas como la cifoescoliosis, pueden ocasionar trastornos hemodinámicos a mediano-largo plazo; contrariamente otras, como el denominado pectum excavatum, dificultan el examen físico, producen falsa impresión de cardiomegalia, pero casi nunca originan alteraciones cardíacas de significación.
La inspección del aparato cardiovascular no puede considerarse completa si no se exploran detenidamente las venas. El pulso de las venas centrales, que se trasmite hacia la periferia en sentido opuesto a la dirección de la sangre, se debe a los cambios de flujo y presión que produce el llenado intermitente de la aurícula y ventrículo. Por consiguiente, los latidos que se observan en las venas del cuello reflejan la actividad auricular y ventricular derechas y su estudio constituye un método clínico que nos permite analizar la dinámica del corazón derecho al que, con frecuencia, no se le presta toda la atención que merece.
La exploración de las venas del cuello tiene 2 objetivos principales: el estudio de las ondas del pulso venoso y la estimación de la presión venosa. Desafortunadamente, el pulso venoso yugular resulta difícil de examinar en niños pequeños. Para llevar a cabo esta exploración es necesario examinar las 2 venas yugulares, interna y externa, a ambos lados del cuello; en la mayor parte de los pacientes la vena idónea es la yugular interna derecha, que permite una evaluación más precisa de las ondas del pulso. La vena yugular interna está situada debajo del músculo esternocleidomastoideo y al lado de la arteria carótida, por lo que las pulsaciones venosas visibles se deben a la trasmisión al tejido subcutáneo de los cambios de presión-volumen que ocurren en el interior de la vena.
Al realizar la exploración es importante que el enfermo esté en la posición adecuada. Debe colocarse una pequeña almohada debajo del cuello para relajar sus músculos (en particular el esternocleidomastoideo), la cabeza no debe estar rotada, ya que ello puede ocasionar tensión muscular y dificultar la trasmisión de las pulsaciones venosas; el tronco debe incorporarse gradualmente hasta que las pulsaciones adquieran la mayor intensidad (la máxima pulsación de la vena yugular interna se observa cuando el tronco se incorpora entre 15 y 30°). Se facilita el examen si iluminamos tangencialmente la piel que recubre la vena, de esta forma se logra observar la sombra que proyecta el latido venoso en la almohada o en las sábanas de la cama.
No debe confundirse el pulso venoso con el latido carotídeo:
- Las ondas del latido venoso son más lentas y sinuosas que las del pulso arterial, más bruscas y fuertes.
- Las pulsaciones de la vena yugular interna pueden suprimirse comprimiendo suavemente por debajo del punto de observación.
- La respiración puede producir importantes cambios en el pulso venoso, mientras que el pulso arterial se modifica muy poco.
- La compresión abdominal resulta útil para diferenciar el pulso venoso del arterial. Normalmente esta maniobra no modifica el latido arterial y produce solo un ligero aumento de las pulsaciones venosas yugulares. En cambio, cuando existe insuficiencia cardíaca derecha el aumento de la distensión y de los latidos yugulares resulta muy evidente; en estos casos la compresión abdominal aumenta el retorno venoso al corazón que, al ser incapaz de aumentar el gasto cardíaco, provoca su retención en el sistema venoso yugular y aumenta su presión y volumen. Esta maniobra, que recibe el nombre de prueba del reflujo hepatoyugular, es útil para diagnosticar la insuficiencia cardíaca latente.
La exploración correcta del pulso venoso debe hacerse relacionándolo con la auscultación de los ruidos cardíacos.
El pulso venoso normal muestra 3 ondas positivas (a, c y v) y 2 ondas negativas (x, y) (Figura 16):
- Onda a: se debe a la trasmisión retrógrada de la onda del pulso que produce la contracción auricular.
- Onda c: se debe a la trasmisión retrógrada de la onda producida por la contracción ventricular y el abombamiento de la válvula tricúspide en el interior de la aurícula derecha. Comienza normalmente al final del primer ruido cardíaco y alcanza su máxima amplitud después de este.
- Onda x: onda sistólica negativa que se produce a continuación de la onda c. Se debe a 2 factores: Desplazamiento de la base de los ventrículos, incluida la válvula tricúspide, durante la sístole ventricular, ello produce descenso de la presión auricular derecha; y la relajación auricular.
- Onda v: onda positiva telesistólica. Se origina debido a que, al estar cerrada la válvula tricúspide, la aurícula derecha ha aumentado su volumen y presión y a expensas del flujo sanguíneo que ha llegado a ella.
- Onda y: onda negativa ubicada a continuación de la onda v. Se debe a la apertura de la válvula tricúspide y a la entrada en el ventrículo derecho de la sangre procedente de la aurícula y de las venas cavas. La porción inicial del descenso corresponde a la fase de llenado rápido ventricular.

Las alteraciones patológicas de la onda a resultan de interés y se relacionan con el momento de su aparición o su amplitud. La ausencia de onda a puede observarse en la fibrilación auricular y en la taquicardia sinusal; cuando la onda a se define claramente y termina antes del primer ruido cardíaco debe sospecharse la existencia de un bloqueo A-V de primer grado. Las ondas a gigantes indican que la contracción auricular derecha se realiza venciendo una gran resistencia, esta situación puede originarse cuando estamos en presencia de estenosis o atresia tricuspídea, estenosis pulmonar o hipertensión pulmonar. La denominada onda “en cañonazo” resulta de la fusión de una onda a gigante con algunas de las ondas (c, x ó v) del pulso venoso que coinciden con la sístole ventricular (válvula tricúspide cerrada), puede observarse en el ritmo de la unión A-V, extrasístoles auriculares, de la unión A-V o ventriculares, taquicardia ventricular o disociación A-V.
En pacientes pediátricos con patología aórtica se describe el denominado signo de González Sabatier, el cual consiste en la ingurgitación venosa permanente de la vena yugular externa izquierda estando la derecha normal; ello indica compresión del denominado tronco venoso braquiocefálico izquierdo ocasionado por la aorta anómala.
Todas las ondas del pulso venoso suelen ser visibles si la frecuencia cardíaca se encuentra por debajo de 85 a 90 latidos/min; cuando la frecuencia es más elevada suelen fusionarse algunas ondas y su análisis se torna no útil. Como se expresó anteriormente, el pulso venoso yugular es difícil de examinar en los niños pequeños, debido a la frecuencia cardíaca más elevada y a que estos pacientes tienen un cuello más corto.
La exploración de las venas periféricas permite estudiar el nivel de la presión venosa; para ello pueden utilizarse las venas del brazo y del dorso de la mano. Se coloca al paciente semisentado, se sitúa la mano por debajo del nivel del corazón el tiempo suficiente para que se llenen y distiendan las venas del dorso, a continuación se eleva la mano pasivamente, poco a poco, mientras se observan las venas, estas normalmente se colapsan cuando se llega al nivel del ángulo esternal de Louis o al hueco supraesternal. Se considera que la presión venosa está elevada si las venas continúan distendidas por encima de este nivel. La causa más frecuente de elevación de la presión venosa es la insuficiencia cardíaca derecha secundaria a falla ventricular izquierda.
Hay que observar el estado general del paciente: si da la impresión de estar enfermo o tiene aspecto de niño sano. Un buen estado nutricional excluirá la insuficiencia cardiaca en el lactante, ya que la alimentación es el ejercicio más importante que realiza. La hipersudoración puede ser signo de insuficiencia cardiaca.
En el fenotipo se buscan rasgos dismórficos de algún síndrome que con frecuencia se asocian a problemas cardiovasculares, como los síndromes de Down, Williams, Marfan, Turner, Noonan, CATCH 22, Ellis Van Creveld, Holt - Oram, etc.
En el área precordial debe investigarse si presenta latido hiperdinámico y se puede identificar el latido de la punta, que si se encuentra desplazado más allá de la línea medioclavicular izquierda y debajo del 4º espacio intercostal izquierdo indica cardiomegalia, en cuyo caso puede haber incluso asimetría en el tórax con abombamiento del hemitórax izquierdo.
La coloración de la piel puede indicar presencia o ausencia de cianosis. Ésta se define como la presencia de más de 5g de hemoglobina reducida/100 ml de sangre y es detectable clínicamente con saturación de oxígeno inferior al 75% o menor del 85% en el ojo entrenado. La cianosis central, indicativa de cortocircuito derecha - izquierda, está causada por cardiopatías congénitas y problemas pulmonares, detectable en mucosas, especialmente en la lengua. La prueba de hiperoxia permite diferenciar si la causa es cardiológica o respiratoria. Si tras administrar oxígeno al 100% durante 10 minutos la cianosis mejora, la causa será respiratoria, y si no mejora nada o lo hace muy poco, será cardiológica (es indicativo de un cortocircuito derecha - izquierda intracardiaco). La cianosis de larga evolución se asocia con acropaquías o dedos en palillo de tambor (Tabla 2B).
Al estudiar cómo es la respiración, la taquipnea, la disnea, el aleteo o el tiraje pueden ser signos de congestión venosa pulmonar por insuficiencia cardiaca.
En el cuello de los niños mayores se observa el latido carotídeo (visible en caso de pulsos saltones) y la ingurgitación yugular como signo de aumento de presión venosa central.
PALPACIÓN
Debe realizarse la exploración de los pulsos arteriales del paciente. En ellos se valora ritmo, frecuencia, calidad y diferencia entre extremidades. Se palpará el pulso radial (o axilar en lactantes) y el femoral (o pedio en los adolescentes), valorando su calidad graduada de - a +++ (Tabla 2A). Una asimetría en los pulsos, con pulsos normales en las extremidades superiores y disminuidos o abolidos y/o retrasados en las inferiores detecta una coartación de aorta. Es conveniente tomar la presión arterial en las extremidades superior e inferior. Una diferencia mayor de 20 mmHg es patológica.
Mediante la palpación del latido cardiaco se localiza la situación del corazón (izquierda o derecha). Se empieza palpando el área precordial con la palma de la mano, que es más sensible a la vibración, y será fácil detectar frémitos o thrills, que son vibraciones de la pared torácica. Se palpa el ápex con la punta de los dedos para localizar el latido de la punta. En niños se encuentra en la intersección de la línea medioclavicular izquierda con el 4 espacio intercostal izquierdo y es fácil percibirlo en decúbito supino.
La calidad del latido indica el tipo de problema subyacente, de forma que un latido hiperdinámico que levanta la mano manifiesta una sobrecarga de volumen, mientras que un latido sostenido en el que se nota la gran fuerza que ejerce el corazón es debido a una sobrecarga de presión. Un latido quieto puede encontrarse en pacientes con miocardiopatía dilatada. También se palpará el área de la base del corazón, a ambos lados del esternón, así como el hueco supraesternal.
En el abdomen es importante palpar el hígado para identificar su posición, y por lo tanto, el situs del paciente, así como para apreciar su tamaño, ya que la hepatomegalia es el signo más constante (aunque no patognomónico) de la insuficiencia cardiaca en el lactante. Se investigará la presencia de reflejo hepatoyugular (al presionar el hígado se ingurgitan las venas yugulares) y se descartará la existencia de ascitis. Ambos son signos de presión venosa central elevada. Debe palparse el bazo para descartar esplenomegalia, típica de la endocarditis infecciosa.


AUSCULTACIÓN
Precisa de un entrenamiento específico y más difícil de conseguir que otras, más aún en pediatría debido a las altas frecuencias cardiacas del paciente y a su falta de colaboración en numerosas ocasiones.
La detección de un soplo es el principal motivo de derivación a los servicios de cardiología pediátrica, que en gran parte de las ocasiones resulta ser inocente.
Deben auscultarse los pulmones para evaluar la ventilación, y no olvidar auscultar la cabeza, el hígado, la espalda y el epigastrio, donde puede haber malformaciones vasculares que originen un soplo.
La auscultación se realiza con un fonendoscopio de calidad con doble cámara, una campana para los sonidos de baja frecuencia (o graves) y una membrana o diafragma que filtrará estos sonidos de baja frecuencia y dejará oír los de frecuencia más alta (o agudos).
Se deben explorar:
- Foco aórtico: borde derecho del esternón en el 2º espacio intercostal.
- Foco pulmonar: borde izquierdo del esternón en el 2º espacio intercostal.
- Foco tricúspide: a la izquierda del esternón en su parte inferior, en la 5ª articulación condrocostal.
- Foco mitral o apical: en la línea medioclavicular izquierda, en el 5º espacio intercostal.
Se debe estudiar la frecuencia cardiaca y el ritmo. La arritmia sinusal respiratoria es un hallazgo fisiológico en los niños, en la que durante la inspiración aumenta la frecuencia cardiaca, al llegar más sangre al corazón, y durante la espiración disminuye (Figura 17)

RUIDOS Y FASES DEL CICLO CARDIACO
El primer ruido (R1) se debe al cierre de las válvulas auriculoventriculares (mitral y tricúspide) en el inicio de la contracción isométrica ventricular; en ese momento la presión ventricular sobrepasa la auricular. El primer ruido coincide con el pulso arterial (aspecto importante en pediatría para la identificación de los ruidos cardíacos, de acuerdo con la toma simultánea de pulso y auscultación, sobre todo en recién nacidos y lactantes cuyo rango normal de frecuencia cardíaca es mucho más elevado que en otros grupos de edad) y también con la rama descendente de la onda R electrocardiográfica cuando se registran, al unísono, el electrocardiograma y el fonocardiograma. La onomatopeya del primer ruido es “dum”. El desdoblamiento fisiológico del primer ruido se produce por la contracción asincrónica de ambos ventrículos, lo cual trae como consecuencia que el cierre de las válvulas mitral y tricúspide no se produzca simultáneamente. El cierre de la válvula mitral ocurre antes que el cierre tricuspídeo, por lo que aquel constituye el primer componente del primer ruido. El desdoblamiento se ausculta mejor en el foco tricuspídeo, en posición sentado y en espiración, posee la peculiaridad de que no se trasmite hacia la base cardíaca. El desdoblamiento fisiológico del primer ruido se escucha con frecuencia en los niños, adolescentes y adultos jóvenes, cuando es muy exagerado debe sospecharse la existencia de un bloqueo de rama derecha. La intensidad del primer ruido depende de la posición de las valvas de las válvulas AV al comienzo de la sístole ventricular, y en menor grado, de la fuerza de contracción ventricular. El primer ruido reforzado se produce cuando las valvas están ampliamente abiertas en el momento de la sístole ventricular y para cerrarse tienen que recorrer un largo camino. Se origina una prolongación del pase de la corriente sanguínea desde las aurículas hacia los ventrículos en 2 situaciones específicas: cuando el intervalo PR es corto y cuando la presión auricular permanece elevada al final de la diástole, situación esta última que acontece en las estenosis mitral o tricuspídea, en los estados circulatorios hipercinéticos, en la taquicardia, en los cortocircuitos de izquierda a derecha y en las contracciones prematuras ventriculares. El primer ruido débil se debe, contrariamente, a que las valvas A-V flotan casi en oposición en el momento de la sístole ventricular. En la conducción A-V prolongada y el bloqueo A-V de primer grado (PR largo) la contracción auricular se agota antes que sobrevenga la sístole ventricular, y, las valvas, que están juntas, se cierran suavemente. El primer ruido débil puede apreciarse en la carditis reumática activa (debido a bloqueo A-V de primer grado concomitante), en los estados de shock (por la disminución de la presión venosa de llenado), en la miocarditis (por debilitamiento de la fuerza contráctil miocárdica) y en la pericarditis constrictiva o el derrame pericárdico (por ineficiencia hemodinámica telediastólica).
El segundo ruido (R2) se debe al cierre de las válvulas sigmoideas aórtica y pulmonar. Corresponde con el final electrocardiográfico de la onda T y con el final del período de eyección. Resulta de tono ligeramente más alto y más corto que el primer ruido; su onomatopeya es “lop”. En un corazón normal, el cierre de la válvula aórtica ocurre primero que el cierre valvular pulmonar, en todo momento independientemente del ciclo respiratorio, pero el oído humano no es capaz de percibir esta característica y habitualmente solo precisa este desdoblamiento durante la inspiración; ello se debe a que, en la fase inspiratoria, al disminuir la presión intrapleural, se produce un efecto de succión sobre las venas cavas y, de hecho, un aumento del retorno venoso sistémico, lo que trae como consecuencia que el ventrículo derecho maneje, en ese instante, un volumen mayor de sangre que el ventrículo izquierdo y su período de expulsión sea más largo; con ello se retarda el cierre pulmonar y se exagera el asincronismo de cierre de ambas válvulas. Inversamente, en la fase espiratoria se acorta el asincronismo de cierre entre las válvulas aórtica y pulmonar y se deja de oír el desdoblamiento inspiratorio. A todo este suceso se le conoce con el nombre de desdoblamiento inconstante o fisiológico del segundo ruido. El desdoblamiento del segundo ruido auscultable en espiración siempre es patológico (segundo ruido desdoblado constante). Un retraso espiratorio de cierre de la válvula pulmonar puede producirse por:
- Retraso dinámico por aumento electivo del volumen de expulsión del ventrículo derecho; ocurre en los defectos septales interauriculares y en el drenaje anómalo venoso pulmonar.
- Retraso eléctrico de la activación del ventrículo derecho por bloqueo completo de rama derecha del haz de His.
- Retraso mecánico por la prolongación de la sístole mecánica causada por obstrucción en la estenosis pulmonar.
El fonocardiograma permite subdividir al segundo ruido desdoblado constante en fijo o variable de acuerdo con las peculiaridades del desdoblamiento espiratorio. El desdoblamiento invertido o paradójico del segundo ruido (cierre inicial valvular pulmonar y posteriormente aórtico) se puede encontrar en la estenosis aórtica por el retraso mecánico de la sístole ventricular izquierda al luchar contra la obstrucción, en el bloqueo de rama izquierda (retraso eléctrico) y en la persistencia del conducto arterioso por la sobrecarga volumétrica de ventrículo izquierdo (retraso dinámico), aunque en este último caso el desdoblamiento paradójico del segundo ruido no se percibe porque el soplo continuo lo enmascara a la auscultación. El segundo ruido es fuerte en la hipertensión arterial sistémica y pulmonar, en esta última, además, se hace único a la auscultación y su componente pulmonar (cierre valvular pulmonar) puede incluso palparse.
Se encuentra en todos los tipos de hipertensión pulmonar:
- Hipertensión pulmonar hipercinética de los cortocircuitos de izquierda a derecha.
- Hipertensión pulmonar pasiva (venocapilar) de la insuficiencia ventricular izquierda y la estenosis mitral.
- Hipertensión pulmonar reactiva del síndrome de Eisenmenger.
- Hipertensión pulmonar obstructiva de las embolias múltiples.
- Hipertensión pulmonar primaria.
El segundo ruido disminuido se produce debido a causas aórticas, pulmonares o de ambos componentes valvulares. El componente pulmonar del segundo ruido está disminuido en la estenosis pulmonar moderada y ausente en la estenosis pulmonar severa, en la atresia pulmonar y el tronco arterioso. El componente aórtico está disminuido en la estenosis aórtica y en la insuficiencia aórtica grave. Se encuentran ambos componentes disminuidos en el enfisema pulmonar, en la obesidad, en los estados de shock y en el taponamiento cardíaco.
El logro de la auscultación del segundo ruido cardíaco en las 2 fases del ciclo respiratorio resulta fácil en el niño mayor, pero muy dificultoso en edades más tempranas; en niños pequeños es necesario, para ello, realizar compresión manual de fosas nasales y cerrar los labios al mantener en la boca un chupete con jarabe de fresa o similar mientras se ejecuta el proceder.
R1 también puede oirse casi simultáneamente con la palpación del choque del pulso carotídeo, lo que puede ayudar a identificar R1 y R2 en pacientes con frecuencias cardiacas elevadas (recién nacidos y lactante).
El R3 se produce poco después de R2. Se cree que se origina en las vibraciones de la pared ventricular que resultan del impacto de la corriente de sangre que entra durante el llenado ventricular.
El cuarto ruido (R4) es de baja frecuencia, es siempre patológica e implica disfunción diastólica ventricular. Se produce por la contracción auricular al final de la diástole.
Otros ruidos: Chasquidos
Su presencia es siempre patológica:
- Chasquido protosistólico de eyección o apertura. Es un sonido sistólico precoz de alta frecuencia y mínima duración que sigue al primer tono. Se escucha en las estenosis de las válvulas semilunares o en la dilatación de los grandes vasos, como en la hipertensión pulmonar o el truncus arterioso.
- Chasquido mesosistólico, audible en el ápex en caso de prolapso mitral, generalmente seguido de un soplo telesistólico.
- Chasquidos diastólicos de apertura. Se producen después del 2º tono inmediatamente antes de comenzar el llenado ventricular. Se escuchan en la estenosis mitral y tricúspide.
SOPLOS
Son los ruidos producidos por un flujo turbulento en el corazón y los vasos sanguíneos o vibraciones de estructuras cardiacas. Para definirlos, hay que tener en cuenta sus características.
- Fase del ciclo cardiaco en el que ocurren: sistólico, diastólico, sistodiastólico o contínuo. Los soplos sistólicos pueden ser orgánicos o funcionales.
- Duración dentro de la fase del ciclo cardiaco en que sucede el soplo: puede ser precoz (proto-), medio (meso-), tardío (tele-) u ocuparlo todo (holo- o pansistólico) (Figura 18)

- Morfología o forma en que un soplo pasa de la intensidad mínima la máxima o viceversa.
- Eyectivo: in crescendo - decrescendo, romboidal o en diamante. El soplo es creciente hasta alcanzar su intensidad máxima y después decrece progresivamente hasta desaparecer. Es típico de la estenosis de válvulas sigmoideas, audible en la base.
- De regurgitación o pansistólico. La intensidad es constante o rectangular. Es típico de la comunicación interventricular y de la regurgitación de válvulas auriculoventriculares.
- In decrescendo: es el soplo diastólico típico de regurgitación de las válvulas sigmoideas.
- Intensidad sonora: del 1 al 6 mediante la clasificación de Levine (Tabla 3).

- Frecuencia: Alta/agudo, frecuencia media, baja/grave.
- Cualidad o timbre: Puede ser vibratorio o musical, rudo, soplante o aspirativo, en maquinaria, etc.
- Localización: Es el foco o sitio donde es escuchado con mayor intensidad y de forma más nítida.
- Irradiación: Aunque el soplo tenga un punto de máxima intensidad en la auscultación, puede escucharse también en otras áreas, a las que el soplo se propaga de acuerdo con la dirección del flujo de la sangre. Los fenómenos aórticos se irradiarán al cuello, los pulmonares hacia ambos campos pulmonares, los tricúspides al área subxifoidea y el borde esternal derecho bajo, y los mitrales hacia la axila izquierda. Los soplos vasculares de coartación aórtica y conducto arterioso persistente se irradian hacia la espalda.
- Efecto de la respiración: Los soplos originados en el lado derecho del corazón se modifican con la respiración, aumentando con la inspiración.
- Maniobras: Algunas maniobras pueden afectar a las características de los soplos, como el cambio de posición, las maniobras de Valsalva, etc.
Soplos inocentes
Son soplos no patológicos. Es fundamental que el Pediatra los conozca y sepa identificarlos y diferenciarlos de los soplos patológicos, para lo cual deberá tener en cuenta que:
- No se acompañan de síntomas de enfermedad cardiovascular.
- Siempre se acompañan de ruidos cardiacos normales.
- No se acompañan de chasquidos.
- Son sistólicos o contínuos, nunca exclusivamente diastólicos.
- Son de corta duración, nunca holosistólicos. R1 se identificará claramente.
- Muestran una intensidad entre los grados 1-3/6, por lo que no presentan frémito.
- La mayoría de ellos, como los soplos orgánicos, aumentan de intensidad con la presencia de fiebre, anemia o gasto cardiaco elevado.
- Tienen una localización definida, con escasa irradiación.
- Cambian de intensidad al cambiar la posición del paciente.
- Se acompañan de electrocardiograma y radiografía de tórax normales.
Soplos funcionales
- Soplo vibratorio de Still: Más frecuente en niños entre los 2 y 6 años. De tono medio - bajo, de intensidad moderada (grados 2-3/6), confinado a la primera mitad de la sístole y con máxima auscultación en el borde esternal izquierdo y el ápex. Se oye mejor en decúbito y cambia de tono con los cambios posturales. Lo más característico es su tonalidad musical o vibratoria. El diagnóstico diferencial se establece con los soplos de comunicación interventricular, estenosis subaórtica y miocardiopatía hipertrófica, que suelen ser más rudos y asociar frémito.
- Soplo sistólico pulmonar de Fogel: Es audible en el 90% de los niños de 8 a 14 años. Es un soplo eyectivo, romboidal, con un pico de máxima intensidad en el principio o la mitad de la sístole. Es de baja intensidad (1-2/6) y se ausculta en el foco pulmonar. Se oye mejor en decúbito supino y en espiración profunda, y disminuye de intensidad en sedestación o si el niño mantiene el aire al final de la inspiración. Puede ser particularmente intenso en niños delgados, con pectus excavatum o cifoscoliosis, por la proximidad entre el tracto de salida del ventrículo derecho y la pared costal anterior. El diagnóstico diferencial se establece con el soplo que se ausculta en la estenosis pulmonar, que suele ser más intensa, acompañado de un chasquido protosistólico y frémito, y con el de la comunicación interauricular, que se asocia a la palpación de un latido hiperdinámico y a la presencia de un segundo ruido desdoblado amplio y fijo.
- Soplo de estenosis de ramas pulmonares del neonato: Es frecuente en neonatos y lactantes hasta los 6 meses de edad. Está causado por las turbulencias producidas por el tamaño relativamente pequeño de las ramas pulmonares o angulaciones en el origen de las ramas principales de la arteria pulmonar. Es un soplo eyectivo, de intensidad baja (grados 1-2/6) y frecuencia media alta, que se inicia al principio o en la mitad de la sístole y que se extiende hasta el segundo ruido y a veces lo sobrepasa. Se irradia a ambos hemitórax, a las axilas y a la espalda, donde puede oírse mejor en el área precordial. Si persiste más allá de los 6 meses de vida debe investigarse la existencia de estenosis estructurales de las ramas pulmonares, que aparecen en el síndrome de rubéola congénita o en el síndrome de Williams.
- Soplo sistólico supraclavicular o braquiocefálico: Es un soplo eyectivo, romboidal, de máxima intensidad en la zona supraclavicular, que irradia al cuello. Suele oírse con menor fuerza en la parte superior del tórax. Tiene un tono medio - bajo y ocupa la primera mitad o dos tercios de la sístole. Se ausculta con el paciente tanto en decúbito como en sedestación, pero varía con la hiperextensión de los hombros. Debe diferenciarse del soplo de estenosis aórtica, que tiene su máxima intensidad en el foco aórtico y se acompaña de chasquido de eyección y a menudo frémito.
- Soplo sistólico aórtico: Es un soplo sistólico, eyectivo, audible en foco aórtico. Se ausculta ante cualquier afección que aumente el gasto cardiaco, como fiebre, anemia, ansiedad, hipertiroidismo, etc. Debe diferenciarse de los soplos eyectivos por estenosis aórtica o supraórtica y de los soplos de la miocardiopatía hipertrófica con obstrucción dinámica en el tracto de salida del ventrículo izquierdo. Estos últimos aumentan la intensidad con la maniobra de Valsalva y disminuyen al ponerse el niño en cuclillas.
- Zumbido venoso: Es un soplo continuo con mayor intensidad en diástole, que se ausculta con el niño entre los 3 y 6 años de edad. Se oye en sedestación, colocando el fonendoscopio en la parte anterior e inferior del cuello, delante del músculo esternocleidomastoideo, aunque a menudo se extiende a la zona infraclavicular en la pared torácica anterior. Suele ser más intenso en la lado derecho que en el izquierdo. Disminuye o desaparece con la compresión suave de las venas del cuello o al girar la cabeza del niño hacia el lado en el que se ausculta el soplo.
- Soplo de la arteria mamaria: Se ausculta en mujeres durante su último trimestre de embarazo y durante la lactancia, pero también puede oírse en adolescentes. Se inicia en la sístole, aunque puede extenderse hasta la diástole, y es audible en la pared torácica anterior, sobre las mamas. Es un soplo de alta frecuencia y de carácter superficial. Debe diferenciarse del soplo continuo producido por un conducto arterioso persistente o una fístula arteriovenosa.
PRUEBAS DIAGNÓSTICAS BÁSICAS
Radiografía de tórax
Se utiliza en la evaluación iniciar de pacientes con sospecha de cardiopatía hasta disponer de exploraciones más específicas. Puede encontrarse lo siguiente:
- Cardiomegalia: aumento del índice cardiotorácico por encima de 0.5 en niños y 0.6 en lactantes.
- Anomalías en la posición del corazón: un corazón desplazado hacia la derecha (dextrocardia) o en posición centrada (mesocardia), con la punta dirigida a la derecha (dextroápex), junto con la alteración de la posición de la cámara gástrica y la del hígado sugiere una cardiopatía compleja (Figura 19)

- Anomalías en la silueta cardiaca: como la forma de bota en la tetralogía de Fallot, la forma de huevo con pedículo estrecho en la D - transposición de las grandes arterias, o la forma de muñeco de nieve en el drenaje venoso pulmonar anómalo total supracardiaco. Es frecuente la presencia de un cono pulmonar aumentado en cardiopatías con cortocircuito izquierda - derecha, un doble contorno en la zona inferior derecha de la silueta cardiaca en pacientes con dilatación de la aurícula izquierda, o el desplazamiento lateral e inferior de la punta del corazón en las dilataciones del ventrículo izquierdo.
- Anomalías en los vasos pulmonares: en las cardiopatías con flujo pulmonar aumentado suele encontrarse aumento del tamaño de las arterias pulmonares centrales en los hilios, así como en la periferia, con vasos claramente visibles en el tercio lateral de los campos pulmonares, donde no suelen encontrarse normalmente. Pueden encontrarse pulmones negros debido a la escasez de vasos pulmonares en las cardiopatías con flujo pulmonar disminuido.
Electrocardiograma
Es la base para el diagnóstico de arritmias cardiacas. Se debe conocer:
- Frecuencia: Hay que elegir un complejo QRS que coincida con una línea gruesa vertical del papel y contar desde ahí las líneas gruesas hasta el siguiente complejo QRS según la siguiente secuencia numérica: 300, 150, 100, 75, 60, 50. También puede dividirse 60/RR en segundos.
- Ritmo: Lo normal es que el ritmo cardiaco lo marque el nódulo sinusal. En este caso existe una onda P delante de cada complejo QRS que es positiva en DII y aVF y negativa en aVR. Si la onda P es de diferente morfología pero precede a cada complejo QRS, el ritmo provendrá de otro foco en la aurícula. Si no existen ondas P delante de cada complejo QRS, será un ritmo nodal si el complejo QRS es estrecho, o idioventricular si el complejo QRS es ancho. Si las ondas P existen pero no se relacionan con el complejo QRS, se trata de una disociación auriculoventricular.
- Onda P: Además de su eje hay que atender a su duración (menor de 2.5 mm de ancho) y amplitud (menor de 2 - 3 mm de alto). Ondas P anchas se relacionan con crecimiento de la aurícula izquierda, y ondas P picudas, con crecimiento de la aurícula derecha.
- Intervalo PR: Mide el retraso del impulso en el nódulo auriculoventricular. Está alargado en los bloqueos auriculoventriculares y acortado en los síndromes de preexcitación.
- Eje del complejo QRS: Para calcularlo hay que localizar el complejo QRS en DI y aVF, y una vez localizado el cuadrante buscar un complejo que sea isodifásico.
- Análisis del complejo QRS: En el nacimiento predomina el ventrículo derecho sobre el izquierdo. Entre el primer mes de vida y los 3 años se produce la transición, con aumento progresivo de la masa del ventrículo izquierdo. A partir de los 3 años el ventrículo izquierdo domina sobre el derecho. El predominio del ventrículo derecho se manifiesta por ondas R de mayor tamaño en V1 y V2, así como ondas S profundas en V5 y V6. El predominio del ventrículo izquierdo se manifiesta por ondas R altas en DII, V5 y V6 acompañadas de ondas S profundas en V1 y V2.
- Ondas Q: Casi siempre están presentes en V5 y V6, pueden aparecer en DI, DII, DIII y aVF y son patológicas en V1. Suelen tener una amplitud menor de 5 mm y una duración de hasta 0.03 segundos.
- Segmento ST: Suele ser isoeléctrico. Ascensos o descensos de hasta 1 mm en derivaciones de extremidades o 2 mm en precordiales suelen ser normales. Las elevaciones o descensos del segmento ST anormales se asocian con miocarditis, pericarditis, isquemia miocárdica o alteraciones iónicas.
- Intervalo QT: Representa el tiempo necesario para la despolarización y la repolarización ventricular. Su cálculo precisa ser corregido con la frecuencia cardiaca mediante la fórmula de Bazzet (QTc= QT/√RR). Se considera patológico cuando es >/= 450 mseg en hombres o >/= 460 mseg en mujeres.
Ecocardiograma
El estudio ecocardiográfico proporciona una excelente visualización anatómica del corazón y los grandes vasos, así como una valoración funcional. En casos de mala ventana ecográfica (atrapamiento aéreo, obesidad, cirugía torácica reciente) puede realizarse una ecografía transesofágica (se introduce a través del esófago un transductor de tipo sonda y se visualiza desde un plano posterior al corazón). La ecocardiografía transtorácica presenta tres modos básicos.
Modo M: Se obtiene el corte del corazón a lo largo de una única línea de barrido. La imagen obtenida muestra el movimiento a lo largo del tiempo de todas las estructuras cortadas por esa línea de barrido. Se deriva de un corte paraesternal que atraviesa el ventrículo derecho, el septo interventricular, el ventrículo izquierdo y la pared posterior del ventrículo izquierdo. Con este plano pueden medirse con bastante exactitud las dimensiones de los ventrículos y los espesores de las paredes cardiacas y compararlos con las tablas de valores normales para saber su un ventrículo está o no dilatado o diagnosticar la presencia de hipertrofia ventricular. Otro corte en este mismo plano a la altura de la raíz aórtica y la aurícula izquierda permitirá calcular las dimensiones de ambas estructuras.
Modo bidimensional: Se realiza mediante el análisis segmentario del corazón. Los segmentos que se han de analizar son los siguientes: situs visceroauricular, drenajes venosos pulmonar y sistémico, aurículas, tabique interauricular, válvulas auriculoventriculares, ventrículos, tabique interventricular, tractos de salida, válvulas sigmoideas, grandes vasos, coronarias y pericardio. Los planos que utiliza son los siguientes: (Figura 20)

- Plano esternal: Eje largo. Visualización del ventrículo izquierdo, las válvulas mitral y aórtica, el tabique interventricular y el tracto de salida del ventrículo izquierdo.
- Plano paraesternal: Eje corto. Permite visualizar la válvula aórtica cortada transversalmente y valorar el número y la apertura de sus valvas, las arterias coronarias y, a su izquierda, la válvula pulmonar, el tronco y las ramas pulmonares. Con un giro antihorario desde este plano puede obtenerse el plano ductal, para valorar la persistencia de esta estructura. Con el desplazamiento hacia el ápex desde el plano inicial se van cortando transversalmente ambos ventrículos, el tabique interventricular, la válvula mitral y los músculos papilares del ventrículo izquierdo.
- Plano apical de 4 cámaras: Se obtiene una visualización simultánea de las 4 cámaras cardiacas que permite una valoración de su tamaño relativo respecto a las otras y, en el caso de las aurículas, la medición de su área y su comparación con los valores de referencia. También permite valorar las válvulas tricúspide y mitral y el tabique interventricular para diagnosticar la presencia de defectos. Por último, con una angulación anterior del transductor pueden visualizarse los tractos de salida de ambos ventrículos, las válvulas sigmoideas y la primera porción de las grandes arterias.
- Plano subcostal: Para el diagnóstico de los defectos del tabique interauricular. Permite la correcta identificación del situs visceroauricular y la concordancia auriculoventricular y ventriculoarterial.
- Plano supraesternal: Se emplea fundamentalmente para valorar la aorta ascendente, el arco aórtico, los troncos supraaórticos y la aorta descendente. Un giro en este plano permitirá visualizar las venas pulmonares drenando en la aurícula izquierda.
Doppler
Mediante este modo puede estudiarse la dirección y la velocidad del flujo de la sangre dentro del corazón y de los grandes vasos. Indicado en el estudio del flujo en las válvulas para el diagnóstico de estenosis e insuficiencias y la detección de cortocircuitos anómalos.
Doppler pulsado: Permite conocer la velocidad de la sangre en un punto concreto del corazón y con ello, el punto donde ésta se acelera en caso de estenosis. Es importante a la hora de evaluar la función diastólica ventricular
Doppler continuo: No tiene un límite de velocidad máximo, lo que posibilita cuantificar las velocidades aunque sean muy grandes. Sin embargo, no permite localizar el punto donde se genera estenosis.
Doppler color: Es una variante del Doppler pulsado en la que se asigna un color al flujo de la sangre que codifica su dirección (rojo cuando se acerca al transductor, azul cuando se aleja de él, amarillo cuando se acerca acelerado, turquesa cuando se aleja acelerado).
Doppler tisular: Esta técnica de reciente incorporación estudia la velocidad del movimiento de las paredes del corazón.
PRESENTACIÓN CLÍNICA DE BEBÉS CON ENFERMEDAD CARDIACA GRAVE
En muchos recién nacidos, la enfermedad cardíaca congénita no se sospecha hasta después del nacimiento, cuando presenten uno o más signos o síntomas. El momento de la presentación de los signos o síntomas depende de la severidad de el defecto, en el útero de los efectos del defecto, y las alteraciones en la fisiología cardiovascular durante la circulación de transición (es decir, el cierre del conducto arterioso y la caída de RVP). A pesar de la presencia de muchas formas heterogéneas de enfermedades del corazón, un número sorprendentemente limitado de signos y síntomas están presentes en el neonato.
Los soplos del corazón son un hallazgo frecuente en los recién nacidos. Las estimaciones de la prevalencia de los soplos cardíacos en los recién nacidos van desde 1 % a 70 % dependiendo del estudio. Aunque soplos cardíacos en el período neonatal no indican necesariamente enfermedades del corazón, deben ser cuidadosamente evaluados. La ausencia de un soplo no excluye graves anomalías cardíacas que amenazan la vida. Soplos patológicos tienden a aparecer a edades característicos (por ejemplo, soplos asociados con estenosis de la válvula semilunar y la insuficiencia de la válvula auriculoventricular tienden a notarse muy poco después del nacimiento). Por el contrario, los soplos causados por cortocircuito de izquierda a derecha (PDA, VSD) no pueden ser escuchadas hasta la segunda a la cuarta semana de vida. Por lo tanto, la edad del recién nacido cuando el murmullo se observó por primera vez y el carácter del soplo dan pistas importantes sobre la naturaleza del defecto cardíaco.
La cianosis (una coloración azulada de la piel, las uñas y las membranas mucosas) es uno de los signos de presentación más comunes de la enfermedad cardíaca congénita en el recién nacido. Dependiendo de la tez de la piel subyacente, clínicamente la cianosis no es visible hasta que no haya más de 3-5 g/dl de hemoglobina desaturada. La cianosis central depende tanto de la gravedad de la hipoxemia (que determina el porcentaje de saturación de oxígeno) y la concentración de hemoglobina. La cianosis central debe diferenciarse de acrocianosis, coloración azul de las manos y los pies, lo cual es un hallazgo normal en el recién nacido.
La cianosis en el recién nacido debe ser diferenciado entre causas cardíacas y respiratorias. Trastornos pulmonares causan cianosis en el recién nacido debido a cortocircuito de derecha a izquierda. Diversos grados de hipoxemia manifiestan cianosis y pueden ser el resultado de una enfermedad pulmonar primaria y del sistema nervioso central. La cianosis clínica puede ocurrir sin hipoxemia en un neonato con metahemoglobinemia y policitemia.
La mayoría de los bebés con cianosis por cardiopatías congénitas no tienen dificultad respiratoria (por ejemplo, taquipnea, retracciones intercostales, gruñendo, aleteo nasal, disnea, estertores, cianosis). A menudo el grado de cianosis no es proporcional al grado de dificultad respiratoria evaluada a partir de los exámenes físicos y de Rayos X torácico. Si la cianosis está presente y es causada por un shunt derecha a izquierda fijo (lesión cardíaca), el aumento de oxígeno inspirado tendrá poco efecto sobre los gases de la sangre arterial. Sin embargo, si la cianosis es causada por un defecto difusión en los pulmones (trastorno pulmonar), el grado de cianosis menudo disminuye con el aumento de oxígeno inspirado. La prueba de hiperoxia es beneficioso en la enfermedad respiratoria diferencial ante enfermedad cardíaca cianótica. La prueba de hiperoxia se realiza mediante la obtención de mediciones de gases en sangre arterial (preferiblemente de la arteria radial derecha) cuando el bebé está en aire ambiente y luego, después de que el niño haya estado en oxígeno al 100% durante 5 a 10 minutos. Si el PaO2 es superior a 150 mm Hg, la presencia de un shunt de derecha a izquierda es poco probable que sea causa de la cianosis y la cardiopatía congénita cianótica. La oximetría de pulso no puede ser utilizado para la documentación.
La insuficiencia cardíaca congestiva (ICC ) se produce cuando el corazón no puede satisfacer las demandas metabólicas de los tejidos. Los signos y síntomas de insuficiencia cardíaca congestiva reflejan disminución del gasto cardíaco y la disminución de la perfusión tisular. En las primeras etapas, el recién nacido puede hacer taquipnea y taquicardia con un mayor esfuerzo respiratorio, estertores, retraso del llenado capilar hepatomegalia. El edema causado por Insuficiencia cardiaca rara vez se ve en los recién nacidos. Diaforesis, dificultades para la alimentación y falta de crecimiento es aparente en etapas siguientes. Finalmente, la insuficiencia cardíaca congestiva se puede presentar de forma aguda con colapso cardiorrespiratorio, particularmente con defectos obstructivos. Asfixia en el nacimiento y anemia también deben ser considerados como causas de la insuficiencia cardíaca congestiva en los recién nacidos .
Los síntomas comunes asociados con la insuficiencia cardíaca congestiva se pueden entender utilizando los principios fisiológicos anteriormente descritos, por ejemplo:
- Taquicardia. El corazón intenta compensar la disminución del gasto cardíaco (GC) al aumentar ya sea la frecuencia cardíaca (FC) o el volumen sistólico (VS) (GC = FC × VS). Debido a que el miocardio fetal tiene menos elementos contráctiles y es poco inervado por el sistema nervioso simpático, la capacidad de aumentar el volumen sistólico es muy limitada. Por lo tanto el aumento en el gasto cardíaco se logra principalmente por el aumento de la frecuencia cardíaca.
- Agrandamiento cardíaco. La hipertrofia y dilatación del corazón se producen en respuesta al volumen. Esta presión ampliada es evidente en el examen radiografía de tórax.
- Ritmo de galope. El ritmo de galope es un sonido de llenado anormal causada por la dilatación de los ventrículos. Se escucha como un ritmo de triple en la auscultación.
- Disminución Pulsos periféricos / moteado de las extremidades. La disminución del gasto cardíaco resulta en una redistribución compensatoria del flujo sanguíneo a los tejidos vitales. La perfusión tisular periférica se reduce, lo que resulta en manchas de la piel y disminución de los pulsos.
- Disminución de la producción de orina y edema. La disminución de la perfusión renal resulta de la filtración glomerular disminuida. El cuerpo interpreta esto como una disminución en el volumen intravascular y comienza a iniciar mecanismos compensatorios tales como la vasoconstricción y la retención de líquido y sodio. Los neonatos manifiestan esto como el aumento de peso y edema periorbital.
- Diaforesis. La insuficiencia cardíaca congestiva conduce a un aumento en la tasa metabólica y aumento de la actividad del sistema nervioso autónomo, lo que resulta en diaforesis. Esto es representativa de la mayor carga de trabajo del corazón en el fracaso.
ANEXOS
Anexo 1: RESUMEN DE LOS RUIDOS CARDÍACOS ANÓMALOS
Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.

ANEXO 2: SOPLOS CARDÍACOS INOCENTES MÁS FRECUENTES
Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.

ANEXO 3: SOPLOS CARDÍACOS SISTÓLICOS Y DIASTÓLICOS
Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.

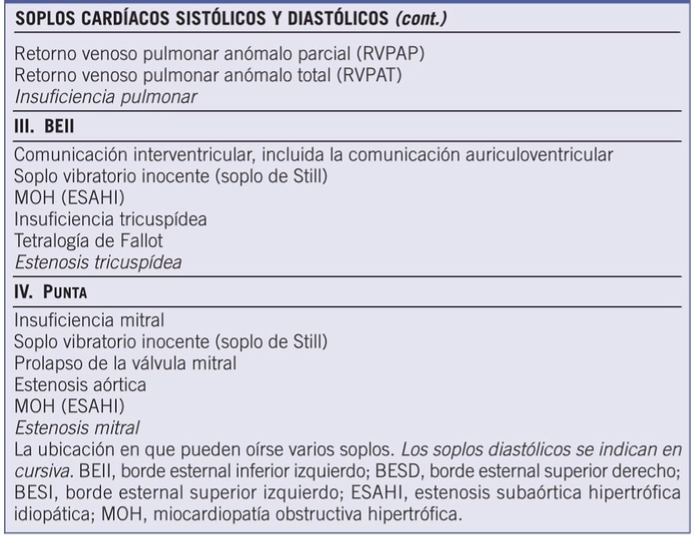
ANEXO 4: ELECTROCARDIOGRAFÍA

Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.
ANEXO 5: Localización de los cuadrantes del eje QRS medio a partir de las derivaciones I y aVF.

ANEXO 6: EJE NORMAL DE LA ONDA T

Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.
ANEXO 7: CRITERIOS DE HIPERTROFIA VENTRICULAR

Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.
ANEXO 8: EFECTOS SISTÉMICOS EN EL ELECTROCARDIOGRAMA


Tomado de: Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.
REFERENCIAS BIBLIOGRÁFICAS
Acerbo F., et al (2009). Cuidados en enfermería neonatal. 3º Edición. Ed. Journal. Hospital de Pediatría SAMIC. Argentina.
Attie F., et al (2012). Cardiología Pediátrica. 2º Edición. Editorial Médica Panamericana. Argentina.
Barret K., et al (2014). Ganong: Fisiología Médica. 23º Edición. Mc Graw - Hill. México.
Carlson B., et al (2012). Embriología humana y biología del desarrollo. 5º Edición. Elsevier Saunders. España
De la Torre E., et al (2006). Pediatría de autores cubanos. Editorial Ciencias Médicas. Habana. Cuba.
Engorn B., et al (2015). Manual Harriet de Pediatría. 20º Edición. Elsevier. España.
Gardner S., et al (2015). Cuidados Intensivos Neonatales. 7º Edición. Editorial Elsevier. USA.
Gleason C., Devaskar S. (2012). Avery: Tratado de enfermedades del recién nacido. 9º Edición. Elsevier Saunders. España.
Gomella T., et al (2013). Neonatología: tratamiento, procedimientos, problemas durante la guardia, enfermedades y fármacos. 6º Edición. Mc Graw Hill. México.
Manotas R. et al (2014). Aspectos Claves en neonatología. 1º Edición. CIB. Colombia.
Hanquinet S., et al (2015). Imagen pediátrica práctica. 1º Edición. Elsevier Saunders. España.
Hoekelman R., et al (2008). Atención Primaria en Pediatría. Ed. Océano Mosby. España.
Lissauer T., et al (2014). Neonatología: lo esencial de un vistazo. 2º Edición. Ed. Médica Panamericana. Argentina.
Mitra, S, Seshan, S, Salcedo, J, & Gil, J 2000, 'Maternal cocaine abuse and fetal renal arteries: a morphometric study', Pediatric Nephrology (Berlin, Germany), 14, 4, pp. 315-318, MEDLINE Complete, EBSCOhost, viewed 10 June 2015.
Moro M., et al (2014). Cruz: Tratado de Pediatría. Asociación Española de Pediatría. 11º Edición. Ed. Médica Panamericana. España.
Moritz, K, Mazzuca, M, Siebel, A, Mibus, A, Arena, D, Tare, M, Owens, J, & Wlodek, M 2009, 'Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats', The Journal Of Physiology, 587, Pt 11, pp. 2635-2646, MEDLINE Complete, EBSCOhost, viewed 10 June 2015.
Navarro-Vargas, J, & Fuentesb, S 2014, 'Monitoría fetal intraquirúrgica: el feto como órgano blanco. (Spanish)', Colombian Journal Of Anesthesiology / Revista Colombiana De Anestesiología, 42, 2, pp. 117-119, MedicLatina, EBSCOhost, viewed 10 June 2015.
Plascencia Ma., et al (2012). Cuidados avanzados en el neonato. Instituto Nacional de Perinatología. Ed. Intersistemas. México.
Polin R. et al (2014). Fisiología Fetal y Neonatal. 4º Edición. Elsevier Saunders. España.
Rentería A., et al (2013). Pediatría de Martínez. Salud y Enfermedad del niño y del adolescente. 7º Edición. Ed. Manual Moderno. México.
Sinha S., et al (2015). Principios de Medicina Neonatal. 5º Edición. Editorial WB. USA.
Tamez S., et al (2012). Enfermería en la unidad de cuidados intensivos. 3º Edición. Editorial Médica Panamericana. Argentina.
Verburg, B, Geelhoed, J, Steegers, E, Hofman, A, Moll, H, Witteman, J, & Jaddoe, V 2007, 'Fetal kidney volume and its association with growth and blood flow in fetal life: The Generation R Study', Kidney International, 72, 6, pp. 754-761, MEDLINE Complete, EBSCOhost, viewed 10 June 2015.
Wilson D., y cols (2012). Manual de Enfermería Pediátrica de Wong. 7º Edición. Mc Graw - Hill. México.


